Актуальные вопросы диагностики муковисцидоза
Статьи
![]()
ЖУРНАЛ «ПРАКТИКА ПЕДИАТРА»
Опубликовано в журнале:
«ПРАКТИКА ПЕДИАТРА»; март-аперль; 2015; стр. 20-27.
Е.И. Кондратьева, д. м. н., профессор, В.Д. Шерман, к. м. н., Н.И. Капранов, д. м. н., профессор, Н.Ю. Каширская, д. м. н., профессор, НКО муковисцидоза ФГБНУ «МГНЦ», ГБУЗ «ДГКБ № 13 им. Н.Ф. Филатова ДЗМ», г. Москва
Муковисцидоз (МВ), или кистозный фиброз (cysticfibrosis), — одно из наиболее частых моногенных наследственных заболеваний с полиорганной патологией, резко сокращающее продолжительность и качество жизни пациентов без адекватного комплексного лечения в течение всей жизни. МВ распространен среди населения всей Земли, но наиболее часто поражает европеоидов: в среднем с частотой 1 на 2500-4500 новорожденных. Еще совсем недавно больные муковисцидозом умирали в раннем детском возрасте или даже на первом году жизни от пневмонии и истощения, обусловленными мальабсорбцией.
Ключевые слова: диагностика, генетика, мутации, неонатальный скрининг, потовая проба, эластаза кала.
Key words: cystic fibrosis, diagnosis, genetics, mutation, newborn screening, sweat test, fecal elastase.
Болезнь прежде всего характеризуется повышенной продукцией вязкого бронхиального секрета, частыми легочными инфекциями и обструкцией дыхательных путей. По мере прогрессирования легочной болезни образуются участки ателектазов, развивается эмфизема, постепенно разрушается паренхима легких с развитием бронхоэктазов и участков пневмосклероза, а больной имеет высокий риск погибнуть от легочно-сердечной недостаточности. В финальной стадии заболевания пересадка комплекса «сердце-легкие» остается для больного единственной надеждой. Помимо бронхолегочной системы у большинства больных муковисцидозом поражается поджелудочная железа, при этом это происходит внутриутробно. Недостаточность панкреатических ферментов обусловливает нарушение всасывания жиров и белков, развитие нутритивной недостаточности. В результате больные отстают в росте и страдают гипотрофией. Продукция инсулина также может быть нарушена, что ведет к развитию диабета. К частым осложнениям течения муковисцидоза относят остеопороз, а также жировой гепатоз с переходом в цирроз. При наличии «мягкой» мутации клинические проявления развиваются постепенно, преобладают моносимптомы, диагноз «муковисцидоз» устанавливается поздно или случайно.
Своевременная диагностика муковисцидоза, обеспечивающая в большинстве случаев раннее начало терапии, в том числе на доклиническом этапе, улучшает прогноз заболевания, повышает эффективность лечения, позволяет предупредить развитие тяжелых осложнений, значительного отставания в физическом развитии, а в ряде случаев и необратимых изменений в легких. Ранняя диагностика позволяет семье вовремя решить необходимые вопросы, связанные с рождением здорового ребенка (генетическое консультирование, пренатальная диагностика МВ в последующие беременности).
Диагностика делится на:
1) пренатальную диагностику;
2) диагностику по неонатальному скринингу (до клинических проявлений или при их дебюте);
3) диагностику при клинических проявлениях:
4) диагностику среди родственников больных.
В настоящее время налаживается дородовая диагностика муковисцидоза в перспективных и информативных семьях (Москва, Санкт-Петербург, Уфа, Томск, Красноярск, Ростов-на-Дону, Владивосток и некоторые другие города), что, безусловно, важно для профилактики этой тяжелой патологии. Пренатальная диагностика возможна в виде ДНК-диагностики при проведении амниоцентеза (получение околоплодных вод в ранний срок -13-14 недель и поздний — обычно 16-20 недель беременности) в семье носителей одной мутации гена CFTR и имеющей больного ребенка. Диагноз может быть заподозрен при УЗИ плода внутриутробно при наличии характерной УЗ-характеристики в виде гиперэхогенного кишечника. УЗИ во время беременности рекомендуют в скрининговые сроки: 11-14, 18-21 и 30-34 недели беременности. Обязательно проводят повторное исследование. В 50-78% случаев это состояние будет связано с МВ и проявится мекониальным илеусом. Диагноз в этом случае может быть установлен еще до рождения ребенка. В то же время этот признак не является высокоспецифичным для МВ, может быть транзиторным явлением, а также связанным с другими патологическими состояниями. При этом ДНК-диагностика родителей дает необходимую информацию о наличии мутаций у каждого из родителей и позволяет предполагать заболевание у ребенка при рождении.
Клинические признаки
1. Диагностика классической формы МВ обычно не представляет сложностей. Классический фенотип больного является результатом наличия двух мутантных копий гена муковисцидозного трансмембранного регулятора (CFTR) и характеризуется хронической бактериальной инфекцией дыхательных путей и придаточных пазух носа, стеатореей из-за внешнесекреторной недостаточности поджелудочной железы, мужским бесплодием из-за обструктивной азооспермии, а также повышенной концентрацией хлоридов потовой жидкости.
2. Проблемы диагностики МВ, как правило, связаны с фенотипическим разнообразием его форм, обусловленным генетическим полимор-
В ряде случаев атипичного течения МВ возможна его диагностика во взрослом возрасте. Как правило, в этой группе больных отмечается более мягкое течение болезни в связи с сохранностью функции поджелудочной железы и нетяжелым поражением органов дыхания.
В абсолютном большинстве случаев МВ может быть диагностирован в раннем детском возрасте (в 90% случаев — на первом году жизни). К сожалению, нередки случаи диагностики МВ у взрослых с классическим фенотипом.
Диагностика МВ у носителей «мягких» генотипов (актуально для детей, рожденных до 2006-2007 гг., и взрослых):
В настоящее время выделяют несколько групп риска по МВ.
Основной группой риска по заболеванию в РФ в настоящее время являются новорожденные с неонатальной гипертрипсиногенемией. Учитывая возможность получения ложноотрицательных результатов неонатального скрининга, а также то обстоятельство, что в РФ неонатальный скрининг на МВ проводится с 2006-2007 гг., не теряет своей актуальности анализ групп риска, включающих пациентов с патологией желудочно-кишечного тракта, бронхолегочными нарушениями, патологией других органов и родственников больных МВ (табл. 1).
Таблица 1.
Группы риска для дифференциальной диагностики муковисцидоза
| I. Бронхолегочные нарушения |
| 1. Повторные и рецидивирующие пневмонии с затяжным течением, особенно двусторонние 2. Бронхиальная астма, рефрактерная к традиционной терапии 3. Рецидивирующие бронхиты, бронхиолиты, особенно с высевом Ps. aeruginosa 4. Двусторонние бронхоэктазы |
| II. Изменения со стороны желудочно-кишечного тракта |
| 1. Синдром нарушенного кишечного всасывания неясного генеза 2. Мекониальный илеус и его эквиваленты 3. Гиперэхогенность кишечника плода 4. Желтуха обструктивного типа у новорожденных с затяжным течением 5. Цирроз печени 6. Сахарный диабет 7. Гастроэзофагеальный рефлюкс 8. Выпадение прямой кишки |
| III. Патология со стороны других органов |
| 1. Нарушение роста и развития 2. Задержка полового развития 3. Мужское бесплодие 4. Хронический синусит 5. Полипы носа 6. Электролитные нарушения |
| IV. Члены семей больных муковисцидозом |
Среди клинических проявлений, характерных для МВ, можно выделить высоко-и менее специфичные (табл. 2). Состояния, представленные в левой колонке таблицы, в абсолютном большинстве случаев встречаются у больных МВ. Причиной состояний из правой колонки могут быть другие заболевания, например первичная цилиарная дискинезия, гуморальный иммунодефицит и т. д.
Таблица 2.
Клинические проявления, характерные для МВ
| Высокоспецифичные для МВ | Менее специфичные для МВ |
| Желудочно-кишечные:
|
Желудочно-кишечные:
|
| Со стороны дыхательных путей:
|
Со стороны дыхательных путей:
|
| Другое:
|
Другое:
|
В таблице 3 представлены особенности проявлений МВ в разные возрастные периоды. Знание этих особенностей помогает специалистам, наблюдающим пациента с теми или иными симптомами, включить МВ в перечень заболеваний для дифференциальной диагностики. Особенно это касается детей раннего возраста, когда клиническая картина еще может быть неполной, но на себя будут обращать внимание некоторые проявления, например мекониальный илеус при рождении или синдром потери солей, не имеющий связи с патологией почек. Диагноз в этом случае может быть установлен еще до рождения ребенка. В то же время этот признак не является высоко специфичным для МВ, может быть транзиторным явлением, а также связанным с другими патологическими состояниями.
Таблица 3.
Клинические особенности проявлений МВ в различные возрастные периоды
| 0-2 года | |
|
|
|
| 3-16 лет | |
|
|
|
Диагностические критерии МВ
Для решения проблем диагностики МВ, в том числе и его атипичных форм, были разработаны критерии, согласно которым обязательным для МВ является наличие характерного клинического синдрома плюс доказательство какого-либо нарушения функции хлорного канала.
Учитывая все научные достижения в понимании природы муковисцидоза и МВ-зависимых заболеваний за последние 10 лет, в 2013 году группа экспертов Европейского общества муковисцидоза (European Cystic Fibrosis Society) под руководством Carlo Castellani подготовила новые стандарты диагностики в редакции Alan R. Smyth и Scott Bell (схема).
Схема.
Диагностические критерии муковисцидоза ECFS 2013
| Положительная потовая проба и/или две мутации МВТР, вызывающие МВ (согласно базе CFTR-2) |
И | Неонатальная гипертрипсиногенемия или характерные клинические проявления, такие как диффузные бронхоэктазы, высев из мокроты значимой для МВ патогенной микрофлоры (особенно синегнойной палочки), экзокринная панкреатическая недостаточность, синдром потери солей, обструктивная азооспермия |
Неонатальный скрининг
Проводится на основании Методических рекомендаций по проведению неонатального скрининга в РФ с использованием Европейских рекомендаций по неонатальному скринингу. 90% новорожденных без клинических проявлений муковисцидоза диагноз может быть установлен на основании скрининга в возрасте до 6 недель. В 5-10% случаев возникают трудности с диагностикой муковисцидоза (Cystic Fibrosis Foundation Patient Registry, 2005 Annual Data Report to the Center Directors. Bethesda, MD: CFF).
Проблемы неонатального скрининга:
Потовая проба
Показания:
1. При положительном результате неонатального скрининга (двукратном повышении уровня иммунореактивного трипсиногена в крови в течение первого месяца жизни ребенка).
2. При наличии у пациента каких-либо характерных клинических проявлений МВ.
3. Случаи МВ в семье.
Потовая проба является надежным методом диагностики МВ у 98% больных. Исследование можно проводить всем детям через 48 часов после рождения, хотя у новорожденных могут быть проблемы с набором пота. Несмотря на то, что «золотым стандартом» диагностики МВ считается количественное определение хлоридов в потовой жидкости (классический метод Гибсона — Кука), метод определения проводимости на аппаратах «Макродакт» и «Нанодакт» («Вескор», США) показал хорошую с ним корреляцию в многочисленных исследованиях.
Оценка результата
При положительном результате потовой пробы (хлориды > 60 ммоль/л при классическом методе Гибсона — Кука и/или проводимость > 80 ммоль/л NaCl) диагноз подтверждается.
Генетическое исследование
Генетическое исследование проводится после потовой пробы. Однако в связи с ограниченными возможностями ДНК-диагностики в России данный метод не является обязательным, однако применяется с исследовательской целью и для окончательного подтверждения диагноза.
На первом этапе ДНК-обследования наиболее часто используется панель, включающая 28 мутаций, как наиболее частых в мире, так и специфичных для России: F508del, CFTRdele2,3(21kb), 3849+10kbC>T, W1282X, 2143delT, 2184insA, 1677delTA, N1303K, G542X, R334W, E92K, L138ins, 394delTT, 3821delT, S1196X, 2789+5G>A, G85E, 2183AA>G, 604insA, 621+1G>T, R117H, R347P, R553X, 3667insTCAA, G551D, I507del, 1717-1G>A, 2184delA. По данным лаборатории генетической эпидемиологии ФГБУ «Медико-генетический научный центр» (МГНЦ) РАМН, при использовании данной панели удается обнаружить лишь около 82,5% мутантных аллелей у больных МВ. В случае когда при положительной потовой пробе не будет найдено ни одной мутации гена (что само по себе маловероятно), может потребоваться секвенирование гена МВ, позволяющее идентифицировать примерно 98% мутаций в гене CFTR.
Рекомендации:
1. На основании данных национального регистра больных МВ по ДНК-диагностике гена CFTR установлены особенности характера и частоты мутаций в регионах страны. На основе данных регистра рекомендуется создание региональных рекомендаций по определению мутаций со ссылкой на регистр (последнюю версию).
2. Отсутствие мутациий без проведения секвенирования — недостаточно для исключения МВ.
3. Некоторые мутации МВТР (3849+10 kb C>T) ассоциированы с нормальным или пограничным результатом потового теста.
4. «Мягкие» мутации характеризуются поздним дебютом заболевания, пограничным значением потовых проб, выявляются чаще при секвенировании.
5. Пациенты с пограничными результатами потовых проб (хлориды 30-60 ммоль/л и/или проводимость 50-80 ммоль/л), единственной мутацией гена представляют реальные трудности для диагностики.
Для диагностики МВ или его исключения при пограничных результатах пробы необходимо:
В европейских странах для подтверждения дефекта ионного транспорта применяется метод определения разности назальных потенциалов или измерение электрического тока в биоптате кишки, отражающие нарушение функции хлорного канала. Оба метода основаны на электрическом характере транспорта ионов и являются высокоинформативными для диагностики МВ.
Диагностика панкреатической недостаточности включает:
У больных МВ показатель эластазы может снижаться в течение первых лет жизни, поэтому определяется в динамике. Низкий уровень панкреатической эластазы расценивается как один из признаков МВ. Приблизительно 1% пациентов с МВ имеет пограничный результат потового теста в комплексе с сохранной функцией поджелудочной железы и хроническим бронхитом.
Диагностика хронического бронхолегочного процесса:
В качестве дополнительных диагностических маркеров могут быть использованы азооспермия в постпубертатном возрасте, идентификация МВ-ассоциированных патогенов из респираторного тракта, рентгенологические признаки синусита.
Знание основных симптомов МВ и особенностей его течения в разные возрастные периоды позволяет своевременно заподозрить наличие заболевания и направить пациента для дальнейшего обследования. Нередкие случаи поздней диагностики МВ связаны как с отсутствием у врачей достаточных знаний о заболевании, так и с фенотипическим разнообразием его форм. Ограниченные возможности ДНК-диагностики МВ в России и ее низкая доступность затрудняют и затягивают окончательную верификацию заболевания.
ЛИТЕРАТУРА
1. Муковисцидоз. Под ред. Н.И. Капранова, Н.Ю. Каширской. М.: ИД «МЕДПРАКТИКА-М», 2014, 672 с. ISBN 978-5-98803-314-1
2. Welsh M.J., Ramsey B.W., Accurso F.J., Cutting G.R. Cystic fibrosis. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., eds. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill, 2001: 5121-88.
3. European cystic fibrosis society standards of care working group. Best practice guidelines. В редакции Alan R. Smith и Scott Bell, 2014.
4. Farell P.M., Rosenstein B.J., White T.B. et al. Cystic fibrosis foundation. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report // J. Pediatr., 2008; 153 (2): S4-S14.
5. Красовский С.А., Каширская Н.Ю., Усачева М.В., Амелина Е.Л., Черняк А.В., Науменко Ж.К. Влияние возраста постановки диагноза и начала специфической терапии на основные клинико-лабораторные проявления заболевания у больных муковисцидозом // Вопросы современной педиатрии, 2014, т. 13, № 2, с. 36-43.
6. de Boeck K., Wilschanski M., Castellani C. et al. Cystic fibrosis: terminology and diagnostic algorithms. Thorax, 2006; 61: 627-635.
7. de Oronzo M.A. Hyperechogenic fetal bowel: an ultrasonographic marker for adverse fetal and neonatal outcome? // J. Prenat. Med., 2011 Jan-Mar; 5 (1): 9-13.
8. Bombieri C. et al. Recommendations for the classification of diseases as CFTR-related disorders // Journal of Cystic Fibrosis, 2011, vol. 10, suppl. 2; S86-S102.
9. Hall E., Lapworth R. Use of sweat conductivity measurements. Annals of Clinical Biochemistry, 2010; 47: 390-392.
10. Sands D., Oltarzewski M., Nowakowska A., Zybert K. Bilateral sweat tests with two different methods as a part of cystic fibrosis newborn screening (CF NBS) protocol and additional quality control. Folia Histochem Cystobiol., 2010 Sep 30; 48 (3): 358-65.
11. Sezer R.G., Aydemir G., Akcan A.B. et al. Nanoduct sweat conductivity measurements in 2664 patients: relationship to age, arterial blood gas, serum electrolyte profiles and clinical diagnosis // J. Clin. Med. Res., 2013 Feb; 5 (1): 34-41.
12. Петрова Н.В. Молекулярно-генетические и клинико-генотипические особенности муковисцидоза в российских популяциях. Автореф. дисс. докт. биол. наук. М., 2009, 42 с.
13. Derichs N., Sanz J., Von Kanel T. et al. Intestinal current measurement for diagnostic classification of patients with questionable cystic fibrosis: validation and reference data. Thorax, 2010 Jul; 65 (7): 594-9.
14. Servidoni M.F., Sousa M., Vinagre A.M. et al. Rectal forceps biopsy procedure in cystic fibrosis: technical aspects and patients perspective for clinical trials feasibility. BMC Gastroenterol., 2013 May 20; 13 (1): 91.
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
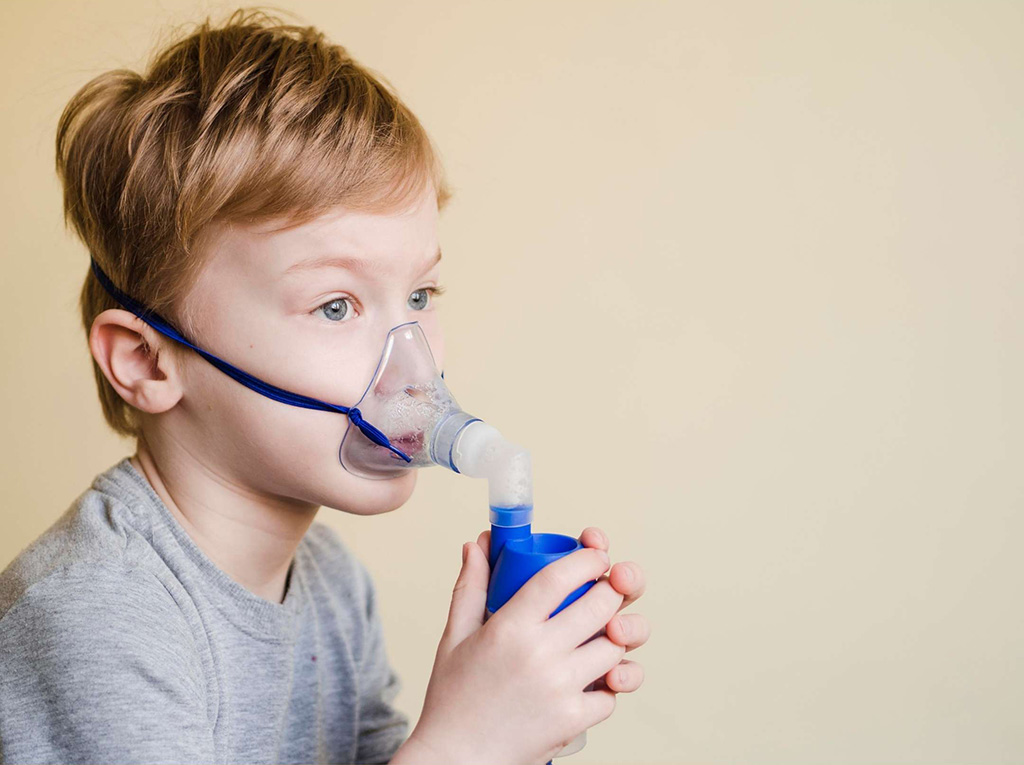
Что нужно знать о муковисцидозе: 5 вопросов о поломке гена, диагностике и лечении
19.04.2022
Муковисцидоз — генетическое заболевание, которое нарушает работу многих органов — легких, кишечника, поджелудочной железы. Из-за этого пациенты часто не могут свободно дышать, используют кислородные маски, ингаляторы, высококалорийные питательные смеси, антибиотики и другие дорогостоящие препараты. Некоторым пациентам требуется пересадка легких.
Какие нарушения происходят в организме при муковисцидозе и какие органы повреждаются чаще всего? Что такое генная терапия и как она может помочь в лечении наследственных заболеваний?
Вместе с информационно-просветительским гуманитарным проектом «12 месяцев», который ведут студенты и ординаторы кафедры патологической анатомии Северо-Западного медицинского университета имени И. И. Мечникова, мы продолжаем серию материалов о редких (орфанных) генетических заболеваниях и жизни людей с ними.
Читайте в апреле рассказ о муковисцидозе, с которым в России рождается один из 10 тысяч детей.
«Уже три года я нахожусь в листе ожидания на пересадку легких»
Лилия, 34 года, подопечная благотворительного фонда «Кислород»:

С раннего детства мне приходилось большую часть своего времени ходить по больницам — в каких только больницах Казани я не была. В 8 лет мне сделали операцию, удалили часть легкого — верхнюю правую долю. Потом оказалось, что это ошибка и операцию не следовало делать. И только в 14 лет мне смогли поставить диагноз «муковисцидоз».
Три года назад я вместе с мамой переехала в Москву и сейчас нахожусь в листе ожидания на пересадку легких. Когда она будет — неизвестно, но мои легкие работают уже всего на 30%.
Возможно, из-за пандемии COVID-19 некоторые люди смогут представить, какого это: ты не можешь пройти даже двух шагов, не начав задыхаться, и потом уже не способен ни о чем думать. Нормальный уровень насыщения крови кислородом 95% и выше, у нас же он всего 60%. Возникает ощущение, будто тебе перекрыли горло и невозможно вдохнуть.
Благодаря фонду «Кислород» мне удается снимать квартиру в Восточном Измайлово, рядом с больницей, в которой мы, пациенты с муковисцидозом, лечимся. Живем около своего доктора.
Что такое муковисцидоз?
Муковисцидоз или кистозный фиброз — тяжелое наследственное заболевание, при котором повышенная вязкость слизи, выделяемая органами пищеварения и дыхания, критически нарушает их работу и приводит к развитию опасных для жизни состояний — истощению, кишечной непроходимости, пневмонии, сепсису, почечной и дыхательной недостаточности. При этом заболевании также часто поражаются печень и сердце, а у мужчин встречается еще и бесплодие.
Считается, что один из 10 тысяч детей в России рождается с муковисцидозом. Сегодня с этим заболеванием в нашей стране живут 3142 человека (по состоянию на 2018 год), а во всем мире около 100 000 человек. Самая высокая средняя продолжительность жизни людей с таким диагнозом — в США и Канаде, она достигает 40 лет.
Муковисцидоз развивается из-за мутаций в гене CFTR (Cystic fibrosis transmembrane conductance regulator). То есть меняется молекула ДНК, на основе которой строится белок — главная рабочая сила клетки. Поломка гена способна полностью прекратить выработку белка или снизить эффективность его работы, в результате чего происходит частичная или полная утрата функции, которую выполняет белок.
Белок, кодируемый геном CFTR, выступает как канал (или отверстие) в мембране (то есть оболочке) клетки, через который из клетки выводятся заряженные частицы — ионы хлора (Cl-). Этот канал получил название «хлоридный» и главное его предназначение — поддерживать в текучем состоянии слизь, которую выделяют наши органы.
Что это за слизь и для чего она нужна? Клетки вырабатывают слизь, покрывающую органы изнутри — например, кишечник, бронхи или протоки поджелудочной железы, — и защищающую их от воздействия ферментов или микроорганизмов. Слизь должна постоянно обновляться за счет удаления из организма, а для этого ей необходимо оставаться текучей — это достигается благодаря тому, что в ней удерживается большое количество воды.
Выделение ионов хлора играет в этом процессе ключевую роль: в сочетании с ионами натрия они формируют и поддерживают в слизи высокую концентрацию соли, а она в свою очередь притягивает в слизь большое количество воды.
У людей с муковисцидозом снижено количество ионов хлора, выводимых в слизь. Это нарушает баланс ионов натрия и хлора и снижает концентрацию соли в слизи. Из-за этого она не снабжается водой, становится вязкой, густой и не может самостоятельно удалиться из организма. Слизь переполняет органы и закрывает просвет выводящих протоков желез, таким образом препятствуя их работе и разрушая структуру. Именно в этом и кроется причина муковисцидоза.
Выявлено уже 352 мутации гена CFTR, которые способны привести к развитию муковисцидоза. Их разделили на 6 функциональных классов:
- I класс — белок не синтезируется;
- II класс — белок не претерпевает нужной обработки в аппарате Гольджи и не может транспортироваться в место функционирования;
- III класс — белок встраивается в место функционирования, но не выполняет свои функции;
- IV класс — белок работает, но проводит недостаточное количество ионов хлора;
- V класс — белок синтезируется в недостаточных количествах;
- VI класс — белок не стабилен и не всегда выполняет свою функцию.

Схема проявлений разных классов мутаций гена CFTR
I, II и III классы относят к «тяжелым» мутациям, при которых функция белка по выведению ионов хлора полностью отсутствует, что ведет к очень низкому содержанию воды в слизи. IV, V и VI классы, при которых функция частично сохранена, — к «легким» мутациям.
Такое подразделение условно, так как на тяжесть течения заболевания значительно влияют два фактора — его раннее выявление и готовность пациента к постоянному лечению. Поздняя диагностика, что часто бывает с легкими мутациями, или прекращение лечения могут привести к необратимым изменениям в организме человека.
Здесь можно узнать, к какому функциональному классу относят болезнетворную мутацию.
«У меня много планов до 27 лет»
Андрей, 24 года, студент Ярославского медицинского университета:

Диагноз «муковисцидоз» мне поставили в 8 месяцев. Позже выяснилось, что у меня 2 тяжелые мутации в гене CFTR. Но до 11 лет болезнь я особенно не замечал, пока не вылетел из школьного обучения на полгода из-за операции по разрешению непроходимости кишечника. Это была моя первая операция, и в этот момент я начал понимать, что болею серьезным неизлечимым заболеванием.
Болезнь начала активно проявлять себя после 17 лет, когда состояние стало резко ухудшаться: появился утренний кашель, часто до тошноты. Муковисцидоз для меня — это постоянное ухудшение состояния легких, кашель, проблемы с пищеварением, сахарный диабет, нарушение работы всех моих органов…
Википедия пишет, что средняя продолжительность жизни с муковисцидозом в России — 27 лет. Классно. Легкие будут все хуже работать, а значит, я не смогу дышать. В один момент могу уснуть и просто не проснуться. Я это понимаю. Поэтому у меня очень много планов до 27!
Как проявляется и на что влияет муковисцидоз?
Мутации гена CFTR способствуют повреждению многих органов — поджелудочной железы, легких, печени, мужских половых желез, слизистых оболочек.

Поражение органов при муковисцидозе
- Поджелудочная железа
Есть однозначная прямая связь между мутациями гена CFTR и поражением поджелудочной железы. Этот орган выполняет две основных функции — вырабатывает пищеварительные ферменты и обеспечивает их выделение в кишечник для переваривания пищи (внешнесекреторная функция), а также производит гормоны и выделяет их в кровь для поддержания уровня глюкозы (внутрисекреторная функция).
При муковисцидозе в первую очередь нарушается внешнесекреторная функция. Как это происходит?
Поджелудочная железа располагается вне кишечника, и для того, чтобы доставить туда пищеварительные ферменты, от разных участков поджелудочной железы к кишечнику отходят маленькие трубочки, которые собираются в крупные выводящие протоки и впадают в кишечник.
Внутренняя поверхность этих выводящих протоков («труб») покрыта специальными клетками, вырабатывающими слизь. Как и в случаях с другими органами, слизь защищает клетки протоков от вредного действия ферментов. Она должна постоянно обновляться, удаляясь из кишечника, но при муковисцидозе становится вязкой, плохо выводится, постепенно закрывая просвет.
Из-за этого ферменты не попадают в кишечник и вместо своей основной задачи по перевариванию пищи начинают «переваривать» клетки поджелудочной железы. Это ведет к ее воспалению — хроническому панкреатиту, который разрушает структуру органа.
Участки с погибшими клетками поджелудочной железы зарастают «рубцами» (фиброз), а продолжающийся процесс выделения слизи расширяет выводящие протоки. В результате в органе появляются полости (кисты), за что заболеванию и было дано первое название — кистозный фиброз.
Нарушенное поступление пищеварительных ферментов в кишечник способствует развитию диареи, плохому перевариванию, истощению, системным нарушениям обмена веществ. Из-за этого пациентам с муковисцидозом приходится постоянно принимать искусственные ферменты.
Потеря внутрисекреторной функции у пациентов с легкими классами мутаций протекает постепенно, на протяжении нескольких лет, а у носителей тяжелого класса нарушения поджелудочной железы выявляются уже внутриутробно. В более позднем возрасте у 20% пациентов развивается инсулин-зависимый сахарный диабет.
Собственные железы кишечника, производящие слизь, также могут поражаться. В совокупности с нарушенным перевариванием пищи это повышает риск развития кишечной непроходимости.
- Легкие
Поражения легких зависят от мутаций гена CFTR, но не столь однозначно, как в случае с поджелудочной железой.
Основной причиной нарушения дыхания считаются микроорганизмы, которые заселяют вязкую слизь дыхательных путей. На инфекцию организм реагирует воспалением: в ткани легкого приходят лейкоциты (а именно нейтрофилы), призванные бороться с микроорганизмами.
Однако при бронхите и пневмонии лейкоциты выделяют ферменты, которые деформируют ткани легкого и способствуют расширению бронхов (бронхоэктазы). Заканчиваются такие разрушительные процессы печально — формируется дыхательная недостаточность, несоответствие количества поглощаемого кислорода потребностям организма.
Из-за этого люди с муковисцидозом не могут свободно дышать, используют кислородные маски, антибиотики и препараты, разжижающие слизь. Они не могут позволить себе легкие физические нагрузки повседневной жизни и встречаться с пациентами с таким же диагнозом, потому что рискуют передать друг другу злостные инфекции.
На течение заболевания легких у пациентов с муковисцидозом влияют гены-модификаторы. Они способны либо ухудшать течение болезни (при особых вариантах генов MBL, HLA II, TGFβ1 функция их белков снижается, что ведет к ослаблению иммунитета и большим повреждениям легких от микроорганизмов), либо наоборот улучшать его (если функциональная активность белка, кодируемого вариантом гена NOS1, повышается, то усиливается антибактериальная защита и ослабляется воспалительная реакция).
- Околоносовые пазухи
Постоянное воспаление характерно и для слизистой оболочки полости носа и придаточных пазух, что часто приводит к образованию доброкачественных опухолей — полипов.
- Сердце
Из-за постоянных пневмоний при муковисцидозе структура легких повреждается, появляются рубцы (фиброз), что ведет к затрудненному кровообращению в них. Это требует напряженной работы сердца — ему приходится с большим усилием проталкивать кровь в сосуды легких. Однако из-за этого развивается гипертрофия правого желудочка, нарушается его снабжение кислородом, что в итоге приводит к сердечной недостаточности.
- Почки
Длительное воспаление в легких стимулирует образование защитных белков, выделяемых в кровь. А так как вся кровь фильтруется через почки, эти белки могут забивать почечные фильтры (амилоидоз почек). Это способствует прогрессированию почечной недостаточности — состоянию, при котором нарушается фильтрация мочи и часть продуктов обмена остается в крови, что имеет критичные и порой смертельные последствия для организма.
- Печень
Поражение печени связано преимущественно с наличием мутаций в генах-модификаторах. Вязкая слизь и густая желчь, образующиеся из-за дефекта гена CFTR, с трудом выводятся в кишечник, способствуя воспалению желчных протоков, гибели печеночных клеток и постепенному формированию цирроза печени.
- Половые органы мужчин
У мужчин с муковисцидозом наблюдается полное закрытие просвета семявыносящих протоков, канальцев придатков яичка (за счет разрастания в них соединительной ткани) или нарушение развития этих органов, что в обоих случаях ведет к бесплодию.
Как наследуется и диагностируется муковисцидоз?
Муковисцидоз наследуется аутосомно-рецессивно: то есть если оба родителя имеют болезнетворную мутацию гена CFTR (считается, что каждый 20-й человек — носитель такой мутации), в 25% случаев может родиться ребенок с муковисцидозом. Причем пол ребенка не влияет на вероятность развития заболевания.
С 2006 года в нашей стране проводится массовое обследование новорожденных на наиболее распространенные наследственные заболевания, среди которых есть и муковисцидоз. К 2018 году почти половину случаев муковисцидоза помог выявить именно неонатальный скрининг. Однако заболевание можно обнаружить и позже из-за его легкого течения. Так, максимальный возраст, в котором был установлен диагноз, в России составляет 59 лет.
Муковисцидоз диагностируется в случае, когда у человека есть как минимум одно клиническое проявление (поражение легких, поджелудочной железы, придаточных пазух носа, кишечника) и подтверждено нарушение функции хлоридного канала.
Нарушение функции канала определяется несколькими подходами:
- Для начала необходимо найти мутацию в гене CFTR и установить ее молекулярно-генетическими методами. Однако наличие мутации еще не говорит о нарушении функции белка.
- Чтобы это подтвердить, используют потовую пробу: с помощью слабого электрического тока (ионофореза) в кожу вводят лекарство, которое стимулирует потоотделение, после чего выделяемый пот собирают и определяют в нем количество хлоридов. Концентрация хлоридов выше 60 ммоль/литр в совокупности с выявленной мутацией гена CFTR свидетельствует о наличии кистозного фиброза у пациента.
История изучения муковисцидоза
Есть ли лечение от муковисцидоза?
Муковисцидоз — неизлечимое заболевание, однако есть способы корректировать его течение, улучшая и продлевая жизнь пациентам.
Основные направления терапии у людей с муковисцидозом:
- Корректировка питания — прием пищеварительных ферментов и использование инсулина при развитии сахарного диабета.
- Корректировка дыхания — использование препаратов, расслабляющих мышцы бронхов и расширяющие их просвет, а также лекарств, разжижающих слизь.
- Антибактериальная терапия при инфекции легких, основная цель которой — сохранить максимально возможный объем функционирующей легочной ткани.
Если функции некоторых органов при муковисцидозе можно скорректировать или заместить приемом лекарств (например, ферментов для пищеварения при повреждении поджелудочной железы), то повреждение легких — это безвозвратный процесс, и компенсировать утрату какой-то их части пока технически невозможно.
Именно критическое поражение легких представляет наибольшую опасность для жизни человека с муковисцидозом.
В таком случае единственно возможным решением остается трансплантация легких. Однако это опасная и рискованная операция, которую делают при снижении функции легких менее 30% от должного уровня.
«Из-за обострений я часто худела до 40 килограммов»
Лилия, 34 года, подопечная благотворительного фонда «Кислород»:

Первый год в Москве я думала: «Ну, мне быстренько сделают пересадку легких, и я уеду домой в Казань». И думала так два года. На третий до меня дошло, что нет, так быстро это не происходит. И у меня была такая истерика, что все вокруг бесило: чужая квартира, чужой город. Готова была выйти на улицу и выть, чуть ли не на стены лезла.
Но я же понимаю, что деваться мне некуда. Когда начинается обострение, начинаешь и реветь, и злиться. От этого же еще хуже, у тебя и так одышка, а когда нервничаешь, одышка усиливается. Это очень тяжело. Успокаиваешь себя: «Все, хватит, перестань, сделаешь себе только хуже». Так сама с собой и разговариваешь.
Хирурги в Москве установили мне трубочку в желудок — гастростому. В нее нужно капать жидкое калорийное питание, чтобы я могла набрать вес и успешнее перенести операцию.
Таким способом я набрала около 10-12 килограммов, а до этого была очень худой, больше 50 килограммов никогда не весила, из-за обострений часто худела и до 40. У нас некоторые и по 35 килограммов весят. Слизь в дыхательных путях очень густая, и ее трудно вывести, нужны большие усилия — мы только от одного кашля огромное число калорий сжигаем.
Месяц назад я начала принимать таргетный препарат «Калидеко» и сейчас чувствую улучшение. Проблема в том, что их не дают бесплатно, если их покупать — курс терапии будет стоить десятки миллионов. Приходится стараться приобретать их самостоятельно, иногда получается достать из-за рубежа.
- Справка: только один из препаратов для пациентов с муковисцидозом входит в программу 14 высокозатратных нозологий и закупается за счет государства. Фонд «Круг добра» закупает такие препараты, но только для детей до 18 лет.
С таргетными препаратами появилась хоть какая-то надежда подольше пожить. Я же осознаю, что далеко не все операции по пересадке легких удачно проходят, 100%-ой гарантии нет. И еще не понимаю полностью, что меня ждет.
Если пересадка легких будет успешной, то я, наверное, на велосипеде покатаюсь, пробегусь. Мы же даже пробежаться не можем, идем тихим-тихим шагом, как черепахи, чтобы не задохнуться. Еще очень хочется попутешествовать. По России я бы поездила, куда угодно — в Сочи, в Петербург….»
Наиболее эффективной медицинской помощью людям с муковисцидозом сегодня остается таргетная терапия, направленная на коррекцию работы хлоридного канала.
В 2012 году был зарегистрирован первый препарат с таким действием — ивакафтор (торговое название «Калидеко»). Он всасывается в желудочно-кишечном тракте и распределяется по организму через кровь.
Достигнув легких, препарат проникает в клетки и присоединяется к хлоридному каналу, встроенному в оболочку (мембрану) клетки. Препарат изменяет форму канала — он возвращает ему способность пропускать ионы хлора, которые способствуют притоку воды в слизь.
По итогам постоянного приема препарата содержание соли в поте пациентов снизилось ниже диагностически значимого уровня, а эффективность работы легких улучшилась на 17,2%. Важный момент: ивакафтор эффективен только при мутациях CFTR, при которых встраивание (транспортировка) белка в оболочку (мембрану) клетки не нарушена, то есть при III-VI классах мутаций.
Позже были разработаны лумакафтор, тезакафтор и элексакафтор — так называемые препараты-корректоры, способные присоединяться к белкам хлоридного канала, что помогает им транспортироваться до оболочки клетки и встраиваться в нее. Они помогают уже и при II классе мутаций.
Препараты-корректоры наиболее эффективны при совместном использовании с ивакафтором, сейчас выпускается несколько таких комбинаций — «Оркамби», «Симдеко» и «Трикафта».
Однако у таргетной терапии есть и недостатки: необходимость принимать препараты каждые 12 часов и их огромная стоимость — цена «Оркамби» или «Трикафта» достигает 20 миллионов рублей в год.
Есть и еще один «минус» — таргетная терапия не способна помочь людям с I классом мутаций, при которых белок (хлоридный канал) вообще не образуется, а следовательно, и воздействовать терапевтической молекуле не на что. Но и для пациентов с этими мутациями ищутся подходы.
«После 17 лет мое здоровье резко пошло под откос, но сейчас животворящий препарат делает свое дело»
Андрей, 24 года, студент Ярославского медицинского университета:

70% лечения муковисцидоза — это подходящая терапия и препараты, еще 30 — массаж, спорт, настрой и правильный уход.
После 17 лет мое здоровье резко пошло под откос, учиться становилось намного труднее. Мне не хватало кислорода, все чаще случались обострения, а в медицинском ВУЗе пропуск учебы равен «смерти».
В итоге из-за болезни я пропустил много занятий, а на их отработку не было ни времени, ни сил. Когда мозг получает мало кислорода, я думаю медленнее, и все тут. Пришлось уйти в академический отпуск и наверстывать упущенное. Но сейчас ситуация улучшается, животворящий препарат от муковисцидоза — «Трикафта» — делает свое дело.
Я принимаю «Трикафту» с сентября 2021 года. И если 2 сентября я только начал ее принимать, то 3 сентября у меня уже прошел кашель. Покашливаю, конечно, но не так, как раньше: у меня были приступы ночного кашля, вплоть до рвоты, и я не мог уснуть до самого утра.
Возможно, если бы я с детства получал таргетный препарат, то не болел бы диабетом, у меня не было бы двух операций и такого резкого ухудшения состояния. Но в то время их еще не было. «Трикафта» была изобретена и зарегистрирована в США только в 2019 году.
За спасительное лекарство пришлось судиться с местным департаментом здравоохранения. «Трикафта» — не зарегистрированный в России препарат. В 2020-м году мама писала запрос о том, что он мне необходим, что требуется провести врачебный консилиум, на что нам ответили только через год: «Нет». Причем это было уже после того, как я попал на консилиум московского центра и получил заключение о необходимости этого препарата для меня…
Мне удалось получить «Трикафту», и это, скорее, исключение из правил, но я не хочу называть это исключением. Я хочу, чтобы все, кто борется за таргетные препараты, получали их. Умирать в 20 лет — это не нормально.
- Справка: Решение о необходимости назначения таргетной терапии принимает консилиум врачей в отношении конкретного пациента. Партии незарегистрированных в России лекарств также завозят на основании заключения, которое Министерство здравоохранения выдает в каждом новом случае. «Сейчас этот алгоритм занимает у пациентов много времени и нередко заканчивается ситуациями, в которых лекарства приходится получать через судебные инстанции», — поясняет фонд «Острова». В январе 2022 года врачи, пациенты и НКО опубликовали письмо с просьбой зарегистрировать препарат «Трикафта» и покупать его взрослым пациентам с муковисцидозом за счет федерального бюджета.
Чем может помочь генная терапия?
Описанные подходы могут лишь скорректировать течение заболевания, но не устранить его причину. Более технологичный подход основан на доставке в клетку здорового гена взамен поврежденному, он получил название «генная терапия».
Подобный принцип уже используется по всему миру, например, при вакцинации от коронавируса. Спутник V представляет собой безвредный вирус — он доставляет в клетку человека ген, с помощью которого производится белок коронавируса. Именно этот белок — чужеродный для организма, не известный ему — стимулирует формирование иммунитета к коронавирусной инфекции, при этом развития COVID-19 не происходит, так как вирус не размножается.
Легкие — основная цель генной терапии, а следовательно, и основное место, куда нужно доставить ген CFTR. Такой подход способен помочь людям с мутациями любых классов, в том числе и в случаях, когда белок не производится клетками.
Для перемещения терапевтического гена в клетку можно использовать различные способы доставки (векторы), например, аденоассоциированный вирус (AAV) и липосомы («пузырьки» из клеточных мембран). После проникновения в клетку терапевтический ген достигает ее ядра, с помощью которого сначала образуется мРНК — информационный посредник между геном и собирающимся белком. Затем белок модифицируется и встраивается в клетку для выполнения функции хлоридного канала.

Основные подходы в генной терапии муковисцидоза. Использование разных способов доставки гена (векторов), например: аденоассоциированный вирус (AAV) и липосомы («пузырьки» из клеточных мембран). Разные механизмы введения вектора: аэрозоли (слева) и внутривенное введение (справа).
Первые попытки разработки и применения генной терапии были осуществлены в 1993 году, но результаты не оправдали ожиданий. Исследователи столкнулись с разными сложностями: как вирусные, так и невирусные векторы могли вызвать воспаление легкого, проникновение вектора в клетки через вдыхание аэрозоля было затруднено из-за наличия вязкой слизи, а при введении через вены вирусы случайным образом распределялись по организму и лишь в редких случаях достигали легких.
Попытки разработать препараты генной терапии не прекращаются:
- Препарат на основе аденоассоциированного вируса. В стадии доклинических исследований (то есть идут испытания на животных) находится терапевтический агент SP-101 (Spirovant, США), в котором здоровый ген доставляет аденоассоциированный вирус. Этот препарат опробован на хорьках с искусственно вызванной мутацией гена CFTR. Исследования показывают эффективность доставки и сохраненную функциональную активность здорового гена.
- Препарат на основе матричной РНК (мРНК) гена CFTR. Альтернативный подход основан на доставке в клетки дыхательных путей матричной РНК (мРНК) гена CFTR. мРНК — информационный посредник между геном и образующимся белком.
Терапевтический агент LUNAR®-CF (Arcturus Therapeutics, США) — мРНК CFTR, помещенная в липосому (пузырек из клеточной мембраны), также проходит этап доклинических исследований. Эффективность доставки и работы препарата также показаны в испытаниях на животных с мутацией CFTR и нечеловекообразных обезьянах.
Основным применением разрабатываемой генной терапии рассматриваются варианты мутации I и II класса, при которых не эффективна таргетная терапия ивакафтором и корректорами хлоридного канала.
Еще одно направление терапии, цель которой — исправить работу гена, основывается на редактировании его структуры. При этом подходе в клетку доставляются молекулярные инструменты, исправляющие поврежденный участок гена. Один из таких препаратов разрабатывается компанией SalioGen (США) и сейчас находится в фазе доклинических исследований.
«В обществе нужно говорить о том, что мы есть и у нас есть право на лекарства»
Андрей, 24 года, студент Ярославского медицинского университета:

Что я хочу сказать миру? Да, мы кашляем, обычно мы очень худые. Но мы абсолютно такие же люди, которые хотят жить и достойны нормальной спокойной жизни. И мы в большинстве своем — дети.
Людям с муковисцидозом хочется сказать, что нельзя опускать руки. Резкое ухудшение состояния у меня началось именно в тот момент, когда я опустил руки. Да, лекарственное обеспечение — это очень важно. Но еще важно не отчаиваться, как бы сложно ни было, постоянно держать себя в узде: принимать препараты с утра и до вечера, делать уколы и ингаляции. Нельзя доводить себя до очень тяжелого состояния, хотя не всегда это от тебя зависит.
Если мне становится совсем сложно, то я вытаскиваю себя спортом, музыкой. Мне повезло: у меня есть девушка, которая меня поддерживает.
Общество может помочь нам распространением информации о муковисцидозе. Нужно говорить о том, что такие люди, как мы, есть, и у нас есть право на лекарства, как бы дорого они не стоили.
Было бы хорошо, если бы людям с таким диагнозом и их близким полагалась обязательная помощь психотерапевта, потому что очень сложно выносить все эти проблемы без поддержки. Очень нужны психологи, чтобы помогать родителям общаться с детьми, принимать их диагноз.
Изначально я поступал в медицинский университет, чтобы пойти в хирургию — общение с хирургами в больнице сильно повлияло на меня (и сериал «Доктор Хаус»). А потом принял решение, что лучше заниматься легкими — в этой отрасли в России нужны свои люди. Те, кто точно знают, что такое муковисцидоз.
Где людям с муковисцидозом искать помощь?
Люди с муковисцидозом не могут полноценно работать из-за тяжести заболевания, а современное лечение — дорогостоящая процедура. В России есть несколько фондов, которые оказывают таким пациентам финансовую, социальную, юридическую помощь и психологическую поддержку, среди них:
- «Кислород»
- «Острова»
- «Во имя жизни»
На сайте фонда «Кислород» можно посмотреть списки и других пациентских организаций и благотворительных фондов, которые помогают людям с муковисцидозом и их семьям.
Статья на конкурс «био/мол/текст»: Муковисцидоз — самое распространенное из моногенетических заболеваний (обусловленных поломкой одного гена). При нем нарушено функционирование белка-переносчика ионов хлора через мембрану клетки — хлорного канала CFTR. Так как этот канал отвечает за нормальную работу эпителия в легких, кишечнике, поджелудочной железе и других органах, его дисфункция приводит к накоплению в этих органах слизи, повышению вероятности инфекций и в конце концов к преждевременной смерти. До последнего времени врачи могли лечить только симптомы муковисцидоза: разжижать слизь, расширять бронхи, снижать воспаление, а также уничтожать бактерий антибиотиками, причем все эти меры почти не продлевали жизнь. Но за последние годы был достигнут невиданный прогресс: средняя продолжительность жизни больных возросла более чем в два раза. В этой статье будет рассказано о препаратах, благодаря которым стал возможен такой успех, об истории их создания и перспективах. На данных примерах читатель также узнает, как происходит современная разработка лекарств.
Описание заболевания
Вначале рассмотрим подробнее, что за болезнь муковисцидоз и почему разработка лекарств против него оказалась таким непростым делом.
Большинство случаев этого страшного заболевания диагностируют в первые годы жизни, потому что муковисцидоз поражает все органы, но особенно — легкие и кишечник. Больные страдают от многочисленных нарушений работы почти всех систем организма: дыхательной, пищеварительной, опорно-двигательной, нервной, сердечно-сосудистой и других. Средняя продолжительность жизни больных составляет 30–40 лет (и сильно зависит от качества ухода), 90% пациентов умирает от легочных осложнений.
Муковисцидоз (или кистозный фиброз, как он называется по-английски) возникает у тех людей, у которых плохо работает или отсутствует белок CFTR. Его название расшифровывается как cystic fibrosis transmembrane conductance regulator, то есть регулятор трансмембранной проводимости при муковисцидозе. Сейчас разберемся, какова роль CFTR в норме и почему его недостаток приводит к таким тяжелым последствиям.
CFTR относится к трансмембранным белкам , которые связывают АТФ и меняют за счет этого свою конформацию. Внутри белка открывается канал, который позволяет ионам хлора выходить из клетки наружу. После гидролиза АТФ канал закрывается (рис. 1).
То есть он пронизывает мембрану клетки насквозь.
АТФ — аденозинтрифосфорная кислота, основная молекула, которая запасает и передает химическую энергию в клетках.

Рисунок 1. Диаграмма предполагаемой структуры белка CFTR в закрытом (слева) и открытом (справа) положениях. Два трансмембранных домена образуют канал. Открытие канала контролируется двумя внутриклеточными доменами (NBD1 и NBD2), которые способны связывать и гидролизовать АТФ (голубой). Регуляторный домен (зеленый) содержит сайты фосфорилирования (Р). Активация канала требует наличия остатка фосфорной кислоты на регуляторном домене. NBD1 и NBD2 связывают и гидролизуют АТФ, вызывая открытие канала путем взаимодействия с трансмембранными доменами. Одна из молекул АТФ остается связанной с NBD1 в течение нескольких минут. За это время происходит несколько циклов открывания-закрывания канала, обусловленных связыванием и гидролизом второй молекулы АТФ [4], [5].
Наличие ионов вблизи поверхности клеток необходимо для поддержания нормального осмотического давления, а это важно для циркуляции жидкости в околоклеточном пространстве. Поэтому постоянный контролируемый поток ионов хлора через мембрану необходим для нормального функционирования эпителия легких, кишечника, протоков поджелудочной железы, яичников, потовых желез.
При муковисцидозе в первую очередь поражаются именно эти органы: в железах образуется густая слизь, которая забивает протоки и мешает нормальной работе органов. А вот почему в легких и кишечнике нарушается работа врожденной иммунной системы, возникает хроническое воспаление и инфекции — стало более-менее понятно совсем недавно. На нынешний момент картина примерно такая: снижение концентрации ионов хлора в околоклеточном пространстве вызывает активацию эпителиального натриевого канала ENaC, который начинает закачивать натрий в клетку. Уменьшение концентрации NaCl возле поверхности клетки вызывает снижение осмотической силы, а, следовательно, количества воды, поступающей к клетке. В случае легких это приводит к осушению воздушных путей и снижению очищающей активности ресничек и слизистой оболочки (рис. 2 во врезке).
Постоянный ток слизи вдоль поверхности воздушных путей очень важен для функционирования защитной системы легких. При этом слизь должна быть достаточно жидкой, чтобы растекаться по поверхности эпителия после секреции, но и достаточно вязкой, чтобы движение ресничек эпителия вызывало ее направленный ток. У больных муковисцидозом слизь содержит слишком мало воды, поэтому она густая и неподвижная, от нее трудно избавиться даже при кашле. Собственно, русское название болезни и происходит от двух латинских слов: mucus («слизь») и viscosus («вязкий»).
Поскольку слизь — это полимерная сетка, образованная белками, она содержит поры. Характерная черта слизи при патологии — слишком мелкий размер пор. В нормальной слизи они имеют диаметр 0,2–1 мкм, а при болезни — менее 0,1 мкм. В результате нейтрофилы — клетки иммунной системы, которые в первую очередь отвечают за защиту от бактерий, — не могут проникнуть изнутри сквозь слой слизи. Бактерии на поверхности воздушных путей размножаются беспрепятственно и вызывают хронические инфекции, которые являются главной причиной смертности при муковисцидозе (80% пациентов) [7]. Более того, на уплотненной слизи, в отличие от обычной, бактерии образуют макроколонии, так называемые биопленки, которые особенно устойчивы к действию иммунной системы и антибиотиков [8].
Из сказанного понятно, что одним из средств улучшить состояние больных муковисцидозом должна быть регидратация легких, которую можно обеспечить с помощью ингаляции гипертонического раствора соли. Это паллиативная мера, не воздействующая на причину болезни, но, тем не менее, она позволяет снизить количество осложнений [6].
До открытия молекулярных причин болезни пациентов лечили только симптоматически — разжижая слизь, применяя антибактериальные, противовоспалительные препараты и физиотерапию. У пациентов с нарушениями ЖКТ и поджелудочной железы эффективна терапия диетами и пищеварительными ферментами. Все эти меры облегчают состояние пациентов, однако настоящий скачок в продлении жизни и улучшении ее качества стал возможным только благодаря открытиям молекулярной биологии и рациональной разработке лекарств [9], [10].
Фокус на CFTR
В 1989 году был найден ген CFTR , кодирующий хлорный канал длиной 1480 аминокислот, и начат поиск мутаций, отвечающих за развитие муковисцидоза. Всего описано более 2000 мутаций в гене CFTR, однако только 250–300 из них приводят к муковисцидозу, а достаточно часто встречается (более чем у 0,1% больных) примерно 20 [11].
Курсивом обозначается ген, а прямым шрифтом — соответствующий ему белок.
Мутации удобно разделить на несколько классов в соответствии с тем, какие последствия они вызывают (рис. 3) [12].

Рисунок 3. Классы мутаций CFTR при муковисцидозе. GA — аппарат Гольджи; ER — эндоплазматический ретикулум. Красный овал — ядро клетки.
[13], рисунок адаптирован
Мутации I класса
Мутации I класса встречаются примерно у 10% пациентов. При них белок CFTR вообще не синтезируется или синтезируется в усеченном виде и сразу деградирует, потому что в гене произошла замена кодирующего аминокислоту кодона на стоп-кодон, или сдвиг рамки считывания, или появился сигнал неправильного сплайсинга. Самая частая мутация — замена глицина-542 на стоп-кодон.
Мутации II класса
Наиболее распространены мутации II класса, вызывающие неправильные сворачивание белка и последующий процессинг клеточными механизмами. Самая частая мутация — F508del (делеция фенилаланина в положении 508 ). 70% пациентов гомозиготны по этой мутации (то есть она присутствует в обеих копиях гена CFTR), а у 90% есть хотя бы один мутантный аллель [14]. У гомозиготных пациентов наблюдается тяжелое течение муковисцидоза, а гетерозиготы по CFTR-F508del, у которых одна из копий гена нормальна, не имеют симптомов болезни. Существует гипотеза, объясняющая стабильность такого тяжелого заболевания в человеческой популяции: у гетерозигот в меньшей степени происходит потеря воды при болезнях, сопровождающихся диареей, например, при холере и брюшном тифе. Соответственно, раньше, когда эти болезни были одной из основных причин смертности, особенно детской, шел отбор на дефектные копии гена [15].
F — обозначение фенилаланина, а del обозначает делецию, то есть отсутствие аминокислоты.
Мутация F508del приводит к тому, что белок неправильно сворачивается и еще в эндоплазматическом ретикулуме не проходит «контроль качества» со стороны клеточных систем и направляется на деградацию, не доходя до плазматической мембраны.
Впрочем, 1% неправильно свернутого CFTR-F508del все же может достигнуть клеточной поверхности, но там он работает с очень низкой эффективностью из-за того, что мутация нарушает правильную подвижность доменов, необходимую для открывания и закрывания канала [19] (см. врезку). Кроме того, в течение 2,5 минут белок удаляется с поверхности в эндосомы, и там снова решается его судьба: он либо возвращается обратно в плазматическую мембрану клетки, либо уничтожается. Понятно, что большинство мутантных молекул будет при этом уничтожено.
Мутации III класса
Мутации III класса встречаются у 4–5% пациентов и приводят к неправильной регуляции открытия ионного канала. Из них наиболее обычная — G551D, то есть замена глицина в 551 положении домена NBD1 на аспарагиновую кислоту. Она приводит к тому, что канал остается преимущественно закрытым. Появление в этом положении остатка аспарагиновой кислоты с отрицательно заряженной боковой цепью —CH2–COO− препятствует связыванию АТФ и сближению доменов NBD1 и NBD2 из-за отталкивания отрицательных зарядов между аспартатом и фосфатными группами АТФ, а также кислотными остатками домена NBD2 [19].
Мутации IV и других классов
Довольно редкие мутации класса IV (в сумме 1,7% пациентов) приводят к недостаточно сильному току ионов хлора через открытый канал CFTR [12]. Как правило, это замены положительно заряженных остатков аргинина в канале на незаряженные остатки. По-видимому, наличие положительных зарядов в канале необходимо для прохождения через него ионов Cl−. Для больных с этими мутациями характерно довольно легкое течение болезни, зачастую без легочных и панкреатических проявлений.
Некоторые исследователи различают также мутации классов V–VI, при которых производится работающий белок, но в недостаточных количествах, или происходит быстрое удаление CFTR с поверхности клеток. У таких пациентов течение болезни также сравнительно легкое [11].
Почему для муковисцидоза — самого распространенного наследственного заболевания — первые препараты, направленные на молекулярную причину болезни, появились только недавно, в 2012 году? Причин несколько: во-первых, сломать проще, чем починить, поэтому среди лекарств гораздо больше ингибиторов, блокаторов, антагонистов, чем активаторов и агонистов. В случае CFTR требуется восстановить неработающую функцию, что гораздо сложнее. Во-вторых, при других наследственных заболеваниях, обусловленных дефектом одного гена (например, гемофилии или болезни Гоше), пациентам, как правило, помогает введение дефектного белка в виде инъекций. В случае муковисцидоза проблема так просто не решается. CFTR — мембранный белок, и если его просто ввести пациенту, он не попадет в мембрану эпителиальных клеток и не будет выполнять там нужные функции. Делаются попытки разработать генную терапию муковисцидоза, например, доставить ген CFTR в клетки с помощью вирусных частиц, но они пока не увенчались успехом.
А вот малые молекулы, которые были разработаны с учетом сведений о структуре и функции CFTR, уже произвели революцию в лечении муковисцидоза, и, надеемся, это только начало. О них мы и поговорим дальше.
«Калидеко»: первая ласточка
Сразу после открытия факта, что муковисцидоз вызван мутациями в гене CFTR, у исследователей возникло желание синтезировать вещества, которые могли бы хоть отчасти скомпенсировать эффект данных мутаций. Довольно быстро стало понятно, что эти вещества будут грубо делиться по своей функции на два типа — корректоры, которые будут повышать количество дефектного белка (например, с мутацией F508del) на поверхности клетки, и потенциаторы, которые будут усиливать активность белка, уже находящегося на поверхности. Наиболее простой мишенью при этом выглядят мутации класса III: при них белок правильно свернут, находится на мембране, но преимущественно в закрытом состоянии. Поэтому с 2000-х годов начали поиск таких соединений, которые бы повышали вероятность его пребывания в открытом состоянии [22].
Первые несколько классов соединений были либо недостаточно активны, либо малоселективны (то есть связывались и с другими белками), либо обладали неподходящими фармакологическими свойствами (плохая растворимость, стабильность, проникновение в ткани). Наконец в 2009 году, после многочисленных раундов поиска оптимальной структуры, была опубликована структура вещества VX-770 (рис. 5), позже получившего название ивакафтор (торговое наименование — «Калидеко», Kalydeco) [23]. Расскажем немного подробнее обо всём пути разработки этого соединения.

Рисунок 5. Химическая структура ивакафтора («Калидеко», VX-770) (а) и генистеина (б)
История разработки началась в 2000 году. В ту пору компания Aurora Biosciences, которая занималась разработкой систем скрининга для Большой фармы, решила инициировать свою собственную разработку и получила грант от Фонда муковисцидоза (Cystic Fibrosis Foundation). В 2001 году Aurora была куплена компанией Vertex Pharmaceuticals, и та продолжила исследования.
Скрининг — в данном случае процесс выбора нужной молекулы из большого множества.
Для начала потребовалось создать систему скрининга, которая бы подходила для отбора веществ, активных в отношении мутантов CFTR. Для первичного отбора использовали клеточную культуру мышиных фибробластов, синтезирующих мутантный CFTR-F508del. Для определения влияния исследуемой молекулы на хлорный канал ее добавляли к клеточной культуре, затем добавляли флуоресцентный маркер, чувствительный к изменениям мембранного потенциала из-за потока ионов хлора и «включали» CFTR, добавляя форсколин, активирующий канал.
Так было протестировано 228 000 соединений [24], многие из которых отдаленно напоминали генистеин — известный потенциатор CFTR, который, однако, не подходит для лекарственного применения из-за низкой активности и быстрой деградации в организме.
В итоге в 2005 году был синтезирован и отобран VX-770 — он оказался в 2000 раз мощнее генистеина и показал самый долгий период полувыведения у крыс среди всех аналогов (9,5 ч.). Дальнейшее изучение VX-770 показало, что он не связывается с основными цитохромами P450 , не проявляет активности в отношении 160 важных рецепторов, в том числе нервной системы, не ингибирует сердечный калиевый канал hERG. Все это косвенно свидетельствует о приемлемой безопасности лекарственного кандидата, по крайней мере, на данной стадии.
Время, за которое концентрация вещества, введенного в организм, падает в два раза.
Цитохромы P450 — белки печени, которые отвечают за метаболизм многих лекарств, причем у разных людей по-разному. Чем меньше лекарство на них влияет, тем меньше вероятность взаимодействия с другими лекарствами, потенциальная токсичность и вариабельность действия.
Затем VX-770 тщательно изучали in vitro (на изолированных клетках) и in vivo (на животных) [22]. Определили фармакокинетические параметры молекулы у мышей, крыс, собак и обезьян, показали пероральную биодоступность у крыс и собак на уровне 40–50% (то есть что почти половина проглоченного препарата попадает в кровь).
Исследования эффективности на животных моделях для ивакафтора не проводили, хотя, как правило, это необходимо для получения разрешения на клинические исследования. Но в данном случае подходящей модели не было, потому что ивакафтор не связывается с мышиным CFTR, а трансгенных мышей с человеческим белком не было на момент выхода препарата в клинические исследования.
Разработка препарата шла невероятно быстро: уже в 2008 году были опубликованы первые клинические данные для ивакафтора, а к моменту регистрации в 2012 году были доступны данные трех клинических исследований. На 2018 год в пяти двойных слепых клинических исследованиях [25], [26] уже приняли участие 342 пациента. Основным показателем, который изучается у больных муковисцидозом, является FEV1 — объем форсированного выдоха (forced expiratory volume) за одну секунду. Причем измеряется не абсолютный объем, а процент от нормы для данного пола, возраста, расы и роста. Другие важные показатели — безопасность препарата, частота легочных обострений (выражается в необходимости приема антибиотиков и госпитализации) и качество жизни (определяется по анкете, которую заполняет пациент).

Рисунок 6. Ожидаемая продолжительность жизни без муковисцидоза, с муковисцидозом при применении ивакафтора и с муковисцидозом без ивакафтора
[29], рисунок адаптирован
Ивакафтор у детей и взрослых с мутацией G551D через 24 недели приема по сравнению с плацебо улучшал FEV1 на 5–10% и снижал риск обострений на 55%. У взрослых (но не у детей) он также повышал качество жизни по сравнению с плацебо. Воздействие препарата на ионный канал подтверждалось снижением концентрации хлорида в потовой жидкости [27], [28]. Затем ивакафтор показал эффективность и при наличии других мутаций класса III [11]. Немаловажно, что препарат оказался очень безопасным — частота побочных эффектов не превосходила плацебо. К сожалению, в России он так до сих пор и не зарегистрирован.
Хотя пока недостаточно данных для того, чтобы говорить о снижении смертности от муковисцидоза под воздействием ивакафтора, но проведенное моделирование показывает, что его влияние на FEV1 способно вылиться в увеличение продолжительности жизни в среднем на 15 лет (рис. 6), а также снизить потребность в трансплантации легких [29].
Такая эффективность дала основание компании Vertex установить на свой препарат очень высокую цену: годовой курс стоит в США $310 000. Недавние исследования, проведенные в США и Великобритании, показали, что такая цена неоправданно высока [30], [31].
Несмотря на то, что ивакафтор оказался настоящим прорывом в терапии муковисцидоза, его недостаток в том, что он помогает всего 4–5% больных — у пациентов с гомозиготной мутацией F508del он оказался неэффективен [34]. Такой факт мог бы показаться странным, если учесть, что ивакафтор был отобран по активности на клетках с CFTR-F508del. Однако не нужно забывать: эти клетки предварительно выдерживались при комнатной температуре, что помогает правильно свернуться даже мутантному белку, поэтому на их поверхности было достаточно CFTR-F508del.
Комбинации
Понятно, что для охвата большинства пациентов надо создавать молекулы, эффективные у пациентов с мутацией F508del (напомним, они называются корректорами). Компания Vertex взялась за разработку таких препаратов одновременно с потенциаторами. После скрининга 164 000 молекул и нескольких раундов оптимизации было синтезировано вещество VX-809, которое в клеточных тестах в семь раз улучшало созревание CFTR-F508del и в пять — его способность переносить ионы хлора [35]. Позже оно получило название люмакафтор (lumacaftor).
Люмакафтор пробовали применять в режиме монотерапии (без сочетания с другими препаратами), но он оказался малоэффективен. Поэтому Vertex провела клинические исследования комбинации люмакафтор/ивакафтор, которая получила название «Оркамби» (Orkambi), и зарегистрировала ее в 2015 году.
Оказалось, что добавление люмакафтора к ивакафтору несколько улучшало течение болезни у пациентов с гомозиготной мутацией F508del, хотя эффект был не таким сильным, как в случае применения ивакафтора у пациентов с CFTR-G551D. FEV1 улучшался примерно на 5% по сравнению с плацебо, примерно в полтора раза снижалось количество обострений, улучшалось качество жизни [31]. Комбинация оказалась не столь безопасной, как ивакафтор в одиночку: небольшая часть пациентов прекратила прием препарата из-за удушья. Тем не менее эта комбинация позволила охватить около 45% пациентов с муковисцидозом.
Однако параллельно обнаружили, что у пациентов с мутацией F508del ивакафтор вообще несколько снижает эффективность корректоров [36], и, следовательно, требуются новые препараты и комбинации, чтобы повысить эффективность терапии и охватить тех пациентов, кому не подходят существующие лекарства. Кроме того, оказалось, что люмакафтор активирует цитохром CYP3A — белок печени, который отвечает за метаболизм ивакафтора, что снижает эффективность последнего [37].
Компания Vertex не собирается останавливаться: в 2018 году она вывела на рынок новую комбинацию — тезакафтор + ивакафтор («Симдеко», Symdeco). Тезакафтор тоже был обнаружен в ходе высокопроизводительного скрининга 150 000 соединений в 2005 году [38]. В сочетании с ивакафтором на клетках эпителия бронхов, взятых у пациентов с гомозиготной мутацией CFTR-F508del, он продемонстрировал повышение транспорта хлорида до 15,7% от нормального. Также было показано, что комбинация увеличивает частоту биения ресничек эпителия [39].
В клинических исследованиях Symdeco оказался более эффективным, чем один ивакафтор, а главное — комбинация оказалась совсем безопасной: в группе препарата наблюдалось даже меньше нежелательных явлений, чем в плацебо-группе [31].
По структуре тезакафтор похож на люмакафтор, и в обоих случаях точно не известно, где именно они связываются с CFTR и каков механизм компенсации мутации F508del. Бесценные данные по трехмерной структуре CFTR были получены относительно молодым методом криоэлектронной микроскопии, за который в 2017 году присудили Нобелевскую премию по химии [40].
Три зарегистрированных препарата компании Vertex охватывают потребности примерно 60–70% пациентов и уже продаются больше, чем на $2 млрд (данные 2017 года). Однако и это еще не все — с помощью препаратов следующих поколений Vertex намеревается охватить более 90% пациентов. В том же 2017 году компания потратила на исследования $1,32 млрд.
Перспективы
Vertex ведет исследования новых, тройных комбинаций, где к комплексу тезакафтора и ивакафтора будет добавляться еще один корректор — VX-659, или VX-445, или VX-152. Эти три молекулы были идентифицированы в ходе скрининга в присутствии тезакафтора. На клетках комбинация каждого из этих веществ увеличивала поток ионов хлора до 68–75% от нормы [42].
Результаты исследования фазы 3 для первой такой комбинации станут известны в конце 2018 года, для второй — в середине 2019. В фазе 2 увеличение FEV1 по сравнению с плацебо для тройной комбинации тезакафтор + ивакафтор + VX-659 составило 13%, что свидетельствует о довольно высокой эффективности комбинации [41].
На более ранней стадии есть у Vertex и совсем новые разработки — в фазе 2 исследуется комбинация потенциатора, корректора и ингибитора ENaC. Разработчики надеются, что, снижая отток ионов натрия с поверхности эпителиальных клеток внутрь, они добьются еще лучшего восстановления слизистого слоя.
Еще одна интересная разработка — дейтерированный ивакафтор, то есть такой, у которого некоторые атомы водорода заменены на более тяжелый изотоп дейтерий (рис. 9). Это не влияет на активность ивакафтора по отношению к CFTR, но делает его более устойчивым к превращениям в организме. В итоге его период полувыведения примерно в полтора раза длиннее, чем у ивакафтора, что позволяет дозировать его один раз в день вместо двух. Сейчас для дейтерированного ивакафтора идут исследования фазы 2 [43].

Рисунок 9. Дейтерирование молекулы увеличивает размер атома водорода и меняет характер ее взаимодействия с некоторыми веществами
На совсем ранней стадии у Vertex есть и генная терапия, использующая систему CRISPR-Cas9 [45], и мРНК, компенсирующая дефекты CFTR. Однако множество провалов в этой области пока не дают основания утверждать, что именно эти подходы сработают.
Заключение
К сожалению, пока в России не зарегистрировано ни одно из новых средств, описанных в данной статье, но, надеемся, ситуация в ближайшие годы изменится.
На примере муковисцидоза интересно проследить взаимовлияние между разными уровнями организации материи: изменение всего нескольких атомов в молекуле белка CFTR отражается на работе клетки, затем ткани, органа и всего организма. А последствием этого является организация социальных структур из сотен людей, которые предпринимают усилия для компенсации дефекта на уровне молекул.
Вылечить муковисцидоз пока невозможно — для этого требуются более совершенные средства генной терапии, чем у нас есть сейчас. Но описанные в этой статье препараты позволяют значительно улучшить жизнь большинства больных. Главная задача — разработка таких средств, которые бы помогали всем пациентам независимо от мутаций CFTR.
- CFTR structure and regulation. CFTR.info;
- Кистозный фиброз (муковисцидоз): микробиологическая диагностика хронической респираторной инфекции. (2018). Минздрав РФ;
- Steven V. Molinski, Vijay M. Shahani, Adithya S. Subramanian, Stephen S. MacKinnon, Geoffrey Woollard, et. al.. (2018). Comprehensive mapping of cystic fibrosis mutations to CFTR protein identifies mutation clusters and molecular docking predicts corrector binding site. Proteins. 86, 833-843;
- David C. Gadsby, Paola Vergani, László Csanády. (2006). The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 440, 477-483;
- Bradley S Quon, Steven M Rowe. (2016). New and emerging targeted therapies for cystic fibrosis. BMJ. i859;
- Mark T. Clunes, Richard C. Boucher. (2007). Cystic fibrosis: the mechanisms of pathogenesis of an inherited lung disorder. Drug Discovery Today: Disease Mechanisms. 4, 63-72;
- H. Matsui, M. W. Verghese, M. Kesimer, U. E. Schwab, S. H. Randell, et. al.. (2005). Reduced Three-Dimensional Motility in Dehydrated Airway Mucus Prevents Neutrophil Capture and Killing Bacteria on Airway Epithelial Surfaces. The Journal of Immunology. 175, 1090-1099;
- Richard C. Boucher. (2007). Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends in Molecular Medicine. 13, 231-240;
- Драг-дизайн: как в современном мире создаются новые лекарства;
- Виртуальные тропы реальных лекарств;
- Isabelle Fajac, Claire E. Wainwright. (2017). New treatments targeting the basic defects in cystic fibrosis. La Presse Médicale. 46, e165-e175;
- Michael J. Welsh, Alan E. Smith. (1993). Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 73, 1251-1254;
- Luigi Maiuri, Valeria Raia, Guido Kroemer. (2017). Strategies for the etiological therapy of cystic fibrosis. Cell Death Differ. 24, 1825-1844;
- Nadia Ameen, Mark Silvis, Neil A. Bradbury. (2007). Endocytic trafficking of CFTR in health and disease. Journal of Cystic Fibrosis. 6, 1-14;
- Xin Meng, Jack Clews, Vasileios Kargas, Xiaomeng Wang, Robert C. Ford. (2017). The cystic fibrosis transmembrane conductance regulator (CFTR) and its stability. Cell. Mol. Life Sci.. 74, 23-38;
- Xin Meng, Jack Clews, Eleanor R. Martin, Anca D. Ciuta, Robert C. Ford. (2018). The structural basis of cystic fibrosis. Biochm. Soc. Trans.. 46, 1093-1098;
- Tzyh-Chang Hwang, Jiunn-Tyng Yeh, Jingyao Zhang, Ying-Chun Yu, Han-I Yeh, Samantha Destefano. (2018). Structural mechanisms of CFTR function and dysfunction. J. Gen. Physiol.. jgp.201711946;
- Diane E. Grove, Meredith F.N. Rosser, Richard L. Watkins, Douglas M. Cyr. (2011). Analysis of CFTR Folding and Degradation in Transiently Transfected Cells. Methods in Molecular Biology. 219-232;
- Xin Meng, Jack Clews, Eleanor R. Martin, Anca D. Ciuta, Robert C. Ford. (2018). The structural basis of cystic fibrosis. Biochm. Soc. Trans.. 46, 1093-1098;
- GWAS и психогенетика: консорциумы в поисках ассоциаций;
- Sang Hyun Lim, Elizabeth-Ann Legere, Jamie Snider, Igor Stagljar. (2018). Recent Progress in CFTR Interactome Mapping and Its Importance for Cystic Fibrosis. Front. Pharmacol.. 8;
- Sabine Hadida, Fredrick Van Goor, Jinglan Zhou, Vijayalaksmi Arumugam, Jason McCartney, et. al.. (2014). Discovery of N-(2,4-Di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide (VX-770, Ivacaftor), a Potent and Orally Bioavailable CFTR Potentiator. J. Med. Chem.. 57, 9776-9795;
- F. Van Goor, S. Hadida, P. D. J. Grootenhuis, B. Burton, D. Cao, et. al.. (2009). Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proceedings of the National Academy of Sciences. 106, 18825-18830;
- Fredrick Van Goor, Kimberly S. Straley, Dong Cao, Jesús González, Sabine Hadida, et. al.. (2006). Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. American Journal of Physiology-Lung Cellular and Molecular Physiology. 290, L1117-L1130;
- С миру по нитке: как соединились компоненты клинического исследования;
- Путь к тысячам аптек начинается с одной молекулы;
- Peter J Barry, Anna L Donaldson, Andrew M Jones. (2018). Ivacaftor for cystic fibrosis. BMJ. k1783;
- Sanjay Patel, Ian P Sinha, Kerry Dwan, Carlos Echevarria, Michael Schechter, Kevin W Southern. (2015). Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database of Systematic Reviews;
- Piyameth Dilokthornsakul, Ryan N. Hansen, Jonathan D. Campbell. (2016). Forecasting US ivacaftor outcomes and cost in cystic fibrosis patients with the G551D mutation. Eur Respir J. 47, 1697-1705;
- Penny Whiting, Maiwenn Al, Laura Burgers, Marie Westwood, Steve Ryder, et. al.. (2014). Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis. Health Technology Assessment. 18;
- Modulator treatments for cystic fibrosis: effectiveness and value. (2018). ICER;
- Laura J. Byrnes, Yingrong Xu, Xiayang Qiu, Justin D. Hall, Graham M. West. (2018). Sites associated with Kalydeco binding on human Cystic Fibrosis Transmembrane Conductance Regulator revealed by Hydrogen/Deuterium Exchange. Sci Rep. 8;
- J. P. Clancy. (2014). CFTR Potentiators: Not an Open and Shut Case. Science Translational Medicine. 6, 246fs27-246fs27;
- Patrick A. Flume, Theodore G. Liou, Drucy S. Borowitz, Haihong Li, Karl Yen, et. al.. (2012). Ivacaftor in Subjects With Cystic Fibrosis Who Are Homozygous for the F508del-CFTR Mutation. Chest. 142, 718-724;
- F. Van Goor, S. Hadida, P. D. J. Grootenhuis, B. Burton, J. H. Stack, et. al.. (2011). Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proceedings of the National Academy of Sciences. 108, 18843-18848;
- D. M. Cholon, N. L. Quinney, M. L. Fulcher, C. R. Esther, J. Das, et. al.. (2014). Potentiator ivacaftor abrogates pharmacological correction of F508 CFTR in cystic fibrosis. Science Translational Medicine. 6, 246ra96-246ra96;
- Susanna A. McColley. (2016). A safety evaluation of ivacaftor for the treatment of cystic fibrosis. Expert Opinion on Drug Safety. 1-7;
- N. Pedemonte. (2005). Small-molecule correctors of defective F508-CFTR cellular processing identified by high-throughput screening. Journal of Clinical Investigation. 115, 2564-2571;
- Marc A. Sala, Manu Jain. (2018). Tezacaftor for the treatment of cystic fibrosis. Expert Review of Respiratory Medicine. 12, 725-732;
- Крупные подробности микроскопического мира: Нобелевская премия по химии 2017;
- Marjolein Mijnders, Bertrand Kleizen, Ineke Braakman. (2017). Correcting CFTR folding defects by small-molecule correctors to cure cystic fibrosis. Current Opinion in Pharmacology. 34, 83-90;
- . (2016). Poster Session Abstracts. Pediatr Pulmonol.. 51, S194-S485;
- Scott L. Harbeson, Adam J. Morgan, Julie F. Liu, Ara M. Aslanian, Sophia Nguyen, et. al.. (2017). Altering Metabolic Profiles of Drugs by Precision Deuteration 2: Discovery of a Deuterated Analog of Ivacaftor with Differentiated Pharmacokinetics for Clinical Development. J Pharmacol Exp Ther. 362, 359-367;
- Стрельцова Ю. (2017). Vertex обзавелась улучшенным «Калидеко» против муковисцидоза. «Мосмедпрепараты».
431 просмотр
5 ноября 2022
Здравствуйте, ребёнку 3 недели, сегодня позвонили из поликлиники,повышение итр(подозрение на муковисцидоз) в 2 раза!!!норма 65,у нас 130!!!я очень переживаю, такое повышение говорит о диагнозе???сегодня взяли повторный анализ крови,пока будет результат, я вся изведусь…Ребёнок на гв,стул частый,после каждого кормления,желто зелёный без запаха сильного,не срыгивает,начиталась, теперь и лоб кажется солоноватый у ребенка…набрал за 3 нед 500 грамм.
На сервисе СпросиВрача доступна бесплатная консультация педиатра онлайн по любой волнующей Вас проблеме. Врачи-эксперты оказывают консультации круглосуточно. Задайте свой вопрос и получите ответ сразу же!

Педиатр
Здравствуйте, нужно набраться терпения , перепроверить ещё раз. Прибавка впринципе нормальная для 3х недель. Стул обильный?
Наталья, 5 ноября 2022
Клиент
Ольга, он после каждого кормления, в среднем около наверное чайной ложки,но у меня и со страшим ребёнком в первые месяцы на гв такой частый стул бвл,поэтому как то не запозртла ничего. СКАЖИТЕ пожалуйста на вашей практике такое повышение может дать ложноположит.результат и часто ли они?
Педиатр
На гв это норма. Вообще при муковисцидозе стул обильный и ребёнок плохо прибавляет в весе. На моей практике был такой случай, когда не подтвердили МВ да.

Педиатр, Дерматолог, Венеролог, Детский
Здравствуйте, Наталья
В данном случае не исключена ошибка, но в любом случае нужно пересдать и ждать результата. Достаточно часто бывает дефект сбора, поэтому раньше времени не переживайте
Наталья, 5 ноября 2022
Клиент
Дарья Николаевна, СКАЖИТЕ пожалуйста на вашей практике такое повышение может дать ложноположит.результат и часто ли они?
Педиатр, Дерматолог, Венеролог, Детский
Да, на моей практике было.
Не очень часто, пару раз точно было

Педиатр
Здравствуйте, нужно дождаться повторного результата, терпения🙏🏻Стул такой частоты, консистенции и цвета в норме у всех младенцев
Наталья, 5 ноября 2022
Клиент
Алина Александровна, СКАЖИТЕ пожалуйста на вашей практике такое повышение может дать ложноположит.результат и часто ли они?
Педиатр
Да, может, бывают ошибки лаборатории. Про частоту таких случаев однозначно нельзя сказать, но раньше времени не переживайте, малыш чувствует беспокойство мамы и тоже может беспокоиться

Педиатр
Здравствуйте!
Делать выводы пока ещё рано. Поэтому и сделали повторный забор крови, чтобы исключить неправильный забор и лабораторные ошибки.
Самое главное сейчас успокоиться и ждать результата. Будем надеяться, что это ошибка.
Стул пенится?
Наталья, 5 ноября 2022
Клиент
Эндже, СКАЖИТЕ пожалуйста на вашей практике такое повышение может дать ложноположит.результат и часто ли они?
Стул не пенится
Педиатр
Положительный результат скрининга говорит о том, что есть подозрение на муковисцидоз.
Это ещё не означает 100% наличие. Часто бывают каждых ложноположительные результаты, 3-6 случаев положительного результата только один оказывается действительно больным муковисцидозом, а оставшиеся — это ложноположительные результаты.
В моей практике ещё не было положительных результатов.
Обычно, для постановки диагноза решающее значение имеет потовый тест,

Педиатр
Здравствуйте) нужно дождаться повторного результата, в лабораториях бывают ошибки.

Педиатр
Здравствуйте Наталья, судя по описанию абсолютно здоровый малыш, набор в весе хороший , надеюсь результат ошибочный, наберитесь терпения не переживайте, будем ждать повторный результат, как придут отпишитесь. Будьте здоровы!
Оцените, насколько были полезны ответы врачей
Проголосовало 0 человек,
средняя оценка 0
Что делать, если я не нашел ответ на свой вопрос?
Если у Вас похожий или аналогичный вопрос, но Вы не нашли на него ответ — получите свою онлайн консультацию врача.
Если Вы хотите получить более подробную консультацию врача и решить проблему быстро и индивидуально — задайте платный вопрос в приватном личном сообщении. Будьте здоровы!

Прочтите и возьмите себе на заметку, особенно если вы молодые люди
В России уже много лет проводится массовое обследование новорожденных для выявления у них нескольких наследственных заболеваний. Такое обследование проводится во многих странах и называется скринингом новорожденных или неонаталъным скринингом.
Целью скрининга новорожденных является, конечно, не само выявление новорожденных с еще не проявившимися наследственными заболеваниями, а их лечение, которое позволяет предотвратить появление клинических симптомов, во многих случаях весьма тяжелых, или даже фатальных. В результате рано начатого и аккуратно проводимого лечения вместо тяжело больных детей, а затем подростков и взрослых, получаются здоровые люди, полноценные члены общества, нередко являющиеся гордостью семьи.
Скрининг новорожденных в России ведется в отношении 5 наследственных и врожденных заболеваний: фенилкетонурии, гипотиреоза, галактоземии, адрено-гениталъного синдрома и муковисцидоза.
ЧТО ТАКОЕ МУКОВИСЦИДОЗ?
Муковисцидоз — это наследственное заболевание, обусловленное изменением (мутацией) в гене, который отвечает за синтез белка, осуществляющего в клетках функцию канала для ионов хлора. Из-за нарушения функции этого канала слизь и другие секреты становятся очень густыми и вязкими в легких, поджелудочной железе и других органах. Это приводит к развитию в легких хронической инфекции, повреждающей легочную ткань; нарушению переваривания пищи, поскольку ферменты поджелудочной железы не могут попасть в кишечник, и другим клиническим проявлениям.
КАК НАСЛЕДУЕТСЯ МУКОВИСЦИДОЗ?
Муковисцидоз наследуется по аутосомно-рецессивному типу, когда больные в семье появляются только в одном поколении. Схема такого наследования приведена на рисунке, на котором изображен фрагмент родословной семьи, в которой родился ребенок, больной муковисцидозом. На родословной мужчины обозначены квадратиком, а женщины — кружочком. Внутри этих квадратиков и кружочков нарисована только одна хромосома (из 23 пар, имеющихся у человека), несущая нормальный или мутантный ген муковисцидоза, который помечен черной точкой.
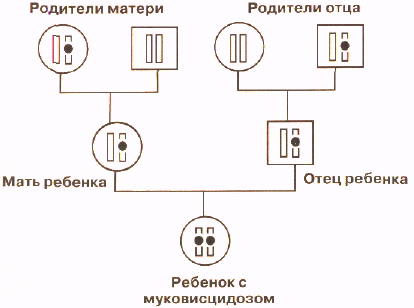
На рисунке для простоты изображена только хромосома, содержащая ген, мутации в котором вызывают муковисцидоз. У ребенка в обеих хромосомах содержится мутантный ген и поэтому он болен. У каждого из родителей мутантный ген содержится только в одной хромосоме, а вторая хромосома нормальная и поэтому они здоровы. Такие люди, которые имеют один нормальный и один дефектный ген называются носителями мутантного гена. У бабки по матери мутантный ген также имеется только в одной хромосоме, как и у деда со стороны отца. Они, как и родители ребенка, здоровы, но передали хромосомы, содержащие мутантный ген, своим детям. У вторых деда и бабки обе хромосомы содержат только нормальный ген. Таким образом, при рецессивном наследовании болен только тот член семьи, который получил от своих родителей обе хромосомы, несущие мутантный ген. Все остальные члены семьи здоровы, в том числе и те, кто является носителем мутантного гена. На схеме родословной видно, что у родителей больного ребенка могут еще появиться больные дети. Вероятность появления больного ребенка в семьях, в которых родители являются носителями мутантного гена, составляет 1/4 или 25%. Эта вероятность не меняется от числа больных или здоровых детей в семье: для каждого следующего ребенка риск, что он будет болен, составляет 25%. Вероятность рождения здорового ребенка, обе хромосомы которого содержат только нормальный ген, составляет также 25%. А 50% детей будут иметь один нормальный и один мутангный ген, как их родители. Это означает, что при каждой беременности родители-носители имеют шансы 3 из 4 (75%) родить здорового ребенка. Многие родители больных муковисцидозом детей и их родственники, первый раз встретившись с врачом-генетиком, настойчиво повторяют, что у их ребенка не наследственное заболевание, так как в их семье ни у кого из родственников никогда не было такого заболевания. Только объяснение, что правила наследования бывают разные, и не редко больной с наследственным заболеванием может быть единственным в семье, позволяют им понять с какой ситуацией они столкнулись.
КАКИЕ ПОРАЖЕНИЯ В ОРГАНИЗМЕ ВЫЗЫВАЕТ МУКОВИСЦИДОЗ?
Заболевание обычно начинается в раннем возрасте. Клиницисты различают три основные формы муковисцидоза: легочную, кишечную и смешанную. Самой частой из них является смешанная форма. Она встречается примерно у 80% больных муковисцидозом. Легочная форма муковисцидоза проявляется хроническим обструктивным бронхолегочным процессом. Из-за того, что мокрота у больных густая и вязкая, она не отхаркивается. Большое количество белка в мокроте делает ее хорошей средой для развития разных микробов, в том числе стафилококков и синегнойной палочки. Развивается хронический воспалительный процесс, приводящий к разрушению легочной ткани. Кровь больных плохо насыщается кислородом, из-за чего начинают страдать сердце, печень и другие . органы. Лечение больных с легочной формой муковисцидоза требует применения мощных антибиотиков в больших дозах. При кишечной форме муковисцидоза нарушается процесс переваривания пищи, так как ферменты поджелудочной железы, расщепляющие белки и жиры, не попадают в кишечник вследствие закупорки протоков железы. Больные отстают от своих родственников в росте и весе. Основное лечение кишечной формы заключается в приеме ферментов поджелудочной железы. Эффективность этого лечения легко контролируется по количеству жира в кале ребенка. Препараты поджелудочной железы должны даваться в таком количестве, чтобы жира в кале не было. При смешанной форме муковисцидоза кишечные проявления муковисцидоза усугубляют поражение легких. Лечение смешанной формы наиболее сложное. У больных муковисцидозом, не получающих необходимого лечения, продолжительность жизни, как правило, относительно короткая.
СУЩЕСТВУЮТ ЛИ ТЕСТЫ НА РАННЕЕ ВЫЯВЛЕНИЕ МУКОВИСЦИДОЗА?
Больные с муковисцидозом могут быть выявлены с помощью скрининга новорожденных. В отличие от других наследственных болезней, выявляемых при скрининге, при которых раннее выявление заболевания позволяет его настолько эффективно лечить, что можно вообще избежать каких-либо клинических проявлений заболеваний, скрининг новорожденных при муковисцидозе преследует несколько иную цель. Врачи считают, что если муковисцидоз выявляется у новорожденного, и его начинают сразу же лечить, то ребенок нормально развивается физически и умственно. У него в меньшей степени будут поражаться легкие и другие органы, увеличится продолжительность жизни, которая в настоящее время, благодаря адекватному лечению, составляет в развитых странах более 35 лет. Скрининг на муковисцидоз начинается с того, что у новорожденного в родильном доме перед выпиской берут из пятки несколько капель крови, которую наносят на специально для этой цели используемую фильтровальную бумагу. Кровь высушивается, и такой бланк, на котором указана фамилия новорожденного и ряд других сведений, необходимых для его идентификации, переправляется в лабораторию региональной медико-генетической консультации. В лаборатории проводят специальное исследование, которое позволяет выявить новорожденных, у которых есть подозрение на муковисцидоз. В этом случае ребенок через педиатра вызывается в лабораторию на повторноетестирование. Родителям педиатр сообщает, что первый тест на муковисцидоз у их ребенка оказался ненормальным. У них появляется повод для беспокойства. Поэтому повторное тестирование образца крови у младенца, которое является очень важным, нужно сделать как можно быстрее. В большинстве случаев при повторном исследовании тест на муковисцидоз оказывается нормальным. Это означает, что результат первого исследования был неверный (его называют ложноположительным). Причины этого могут быть разными и связанными как с состоянием младенца, так и с какой-то ошибкой лаборатории. Этот результат, свидетельствующий о том, что у ребенка нет муковисцидоза, сразу же сообщается родителям, чтобы снять с них чувство страха.
ЧТО ДЕЛАТЬ, ЕСЛИ И ПОВТОРНЫЙ ЛАБОРАТОРНЫЙ ТЕСТ НА МУКОВИСЦИДОЗ ОКАЗАЛСЯ ПОЛОЖИТЕЛЬНЫМ?
Если и второе лабораторное исследование для выявления муковисцидоза оказалось положительным, то, в отличие от других скринируемых наследственных болезней, это еще не означает, что у ребенка есть муковисцидоз, хотя вероятность такого диагноза является высокой. Семья, в которой у ребенка повторно подтвердился положительный тест на муковисцидоз, приглашается на прием к врачу-генетику в медико-генетическую консультацию. Здесь семье объясняют, что собой представляет муковисцидоз и организуют прием у клиницистов, являющихся специалистами в диагностике и лечении муковисцидоза. Теперь наблюдение за ребенком берет на себя группа клиницистов. Для подтверждения диагноза муковисцидоза младенцу проводят так называемый лотовый тест. Это совершенно безобидный и безболезненный тест можно провести младенцу, начиная с возраста 3-4 недели жизни. Если лотовый тест оказывается отрицательным, то ребенок считается здоровым, хотя за ним клиницисты еще будут наблюдать некоторое время. Если же потовый тест оказался положительным, то диагноз муковисцидоза считается установленным, даже до появления каких-либо клинических проявлений заболевания. В этом случае врачи назначат ребенку лечение, которое надо будет строго соблюдать. Ребенок будет периодически обследоваться группой специалистов для постоянного контроля за состоянием его здоровья.
МОЖНО ЛИ ПОМОЧЬ СЕМЬЕ, В КОТОРОЙ ПОЯВИЛСЯ БОЛЬНОЙ С МУКОВИСЦИДОЗОМ, ИМЕТЬ ЗДОРОВЫХ ДЕТЕЙ?
Да, и довольно успешно. Для муковисцидоза возможна дородовая диагностика. Первым шагом в этом направлении является обращение в медико-генетическую консультацию, где врач-генетик определяет показания и возможные методические подходы к дородовой диагностике в каждом конкретном случае. Сама процедура заключается в том, что во время беременности в сроке 9-11 недель или 16-18 недель врач акушер-гинеколог проводит забор очень небольшого количества клеток плода и направляет этот материал в специальную лабораторию пренатальной диагностики. В этой лаборатории врачи лаборанты-генетики проводят молекулярную диагностику, т.е. определяют наличие или отсутствие мутации в гене, отвечающем за муковисцидоз. В случае положительного результата семья решает вопрос о прерывании беременности больным плодом или настраивается на появление еще одного больного ребенка. Это право выбора остается за семьей.
Дата публикации 9 октября 2020Обновлено 26 апреля 2021
Определение болезни. Причины заболевания
Муковисцидоз — это наследственное заболевание, при котором нарушаются функции желёз внешней секреции. Болезнь поражает весь организм, но сильнее всего страдает дыхательная система и поджелудочная железа.
Заболевание также называется кистозным фиброзом, синдромом Фанкони. Характеризуется системным поражением экзокринных желез: слизеообразующих (респираторных, кишечника, поджелудочной) и серозных (слюнных, потовых, слезных).

Частота болезни в России составляет 1:10000 новорождённых [1]. В 10-15 % случаев симптомы муковисцидоза проявляются у ребёнка с первых дней жизни. У таких детей возникает мекониальный илеус, или непроходимость кишечника, при котором его просвет закупорен слишком густым и вязким первородным калом — меконием.
В некоторых случаях симптомы болезни проявляются не сразу. Это связано с тем, что тяжесть и форма заболевания у разных больных отличаются. Кроме того, признаки муковисцидоза могут напоминать симптомы других болезней, что затрудняет диагностику. Без лечения подавляющее большинство больных умирает в детском возрасте, но благодаря современным лекарственным препаратам и другим методам лечения продолжительность жизни пациентов постоянно растёт.
Муковисцидоз — это аутосомно-рецессивное заболевание, то есть болезнь проявляется в том случае, если «дефектный» ген унаследован от обоих родителей. Для этого и отец, и мать должны быть носителями мутации, вероятность рождения больного ребёнка у них составляет 25 % [7].
Дефектный ген муковисцидоза широко распространен — каждый 25-й житель Северной Европы является гетерозиготным носителем мутантного гена. Высокая смертность в сочетании с повышенной фертильностью должны были бы привести к быстром устранению дефектного гена из популяции. Существует теория, по которой ген муковисцдоза сохранился потому, что его носители имели преимущество во время эпидемии холеры — холерная диарея сопровождается потерей натрия и хлора, а при муковисцидозе эти ионы удерживаются в организме [15].

Чаще всего без адекватного лечения болезнь протекает тяжело и имеет плохой прогноз, на который во многом влияет развитие хронической инфекции в нижних дыхательных путях. Бактериальные агенты при муковисцидозе специфичны, например, для раннего возраста характерно развитие инфекции, вызванной золотистым стафилококком, в дальнейшем к нему присоединяются синегнойная и гемофильная палочки. Данные возбудители устойчивы к лекарственным препаратам, что осложняет лечение, поэтому важно своевременно идентифицировать возбудителя и подобрать подходящие антибактериальные средства.
![]()
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы муковисцидоза
При муковисцидозе симптомы болезни зачастую видны сразу после рождения ребёнка и проявляются признаками непроходимости кишечника (мекониальным илеусом):
- выраженное беспокойство и постоянный плач;
- сильное вздутие живота;
- рвота;
- повышенное слюноотделение;
- длительная задержка стула;
- повышенная температура тела;
- частые поносы;
- непереносимость жирной пищи;
- характерный стул — вязкий, липкий, жирный.
Непроходимость кишечника может привести к его перфорации. По статистике, до 70-80 % детей с мекониальным илеусом больны муковисцидозом [3]. Для его исключения каждому новорождённому с кишечной непроходимостью необходимо провести диагностическое обследование.
В возрасте одного года у ребёнка с муковисцидозом может наблюдаться обильный, зловонный стул непереваренной пищей, частый сухой кашель, отставание в физическом развитии и длительная желтуха.
Одним из главных признаков муковисцидоза является повышенная солёность пота. Её замечают родители, когда при усиленном потоотделении на коже ребёнка выделяются крошечные кристаллы соли. Они образуются вследствие повышенной концентрации в потовой жидкости ионов натрия и хлора. Их концентрация увеличивается в результате нарушения транспорта и секреции в клетках, выстилающих потовые железы. При этом потери соли из организма при потоотделении могут превышать её поступление с пищей. При болезни, протекающей с высокой температурой и значительной потерей с потом хлора и натрия, может возникнуть токсикоз и шок [7].
Иногда признаки заболевания проявляются только в школьном возрасте. Дети с муковисцидозом из-за нехватки питательных веществ страдают дефицитом массы тела и отстают в росте.
Для заболевания также характерны:
- изменение стула;
- хроническая диарея;
- вздутие кишечника;
- частые гнойные заболевания лёгких неясной этиологии;
- одышка;
- гаймориты и синуситы.
У заболевших в раннем детстве и доживших до взрослого возраста отмечаются постоянный сухой кашель, вне обострения чаще наблюдается постоянное отделение мокроты от 5-10 мл до 200 мл и более. Характер мокроты — от слизистой до гнойной. Одышка, которая в начале болезни может возникать только при небольшой нагрузке, но со временем усиливается, частые респираторные заболевания и гаймориты. У пациентов выражена задержка роста, фаланги пальцев и ногти изменяются по типу «барабанных палочек» и «часовых стёкол».
Грудная клетка чаще бочкообразной формы. Появляется бледность, одутловатость лица и цианоз видимых слизистых.

У больных может наблюдаться задержка в половом развитии или отсутствие вторичных половых признаков:
- отсутствие волос в подмышечных впадинах и на лобке;
- замедленный рост половых органов;
- недоразвитие молочных желёз у девочек;
- отсутствие менструального цикла.
У женщин с муковисцидозом снижена фертильность, у мужчин наблюдается азооспермия — отсутствие сперматозоидов в эякуляте [3][4].
Патогенез муковисцидоза
Патогенез муковисцидоза обусловлен мутацией гена CFTR (Cystic Fibrosis Transmembrane conductance Regulator) — муковисцидозного трансмембранного регулятора проводимости. Этот ген кодирует белок, который обеспечивает транспорт солей и воды в эпителиальном слое бронхолёгочной системы и в клетках, выстилающих железы, вырабатывающие секрет и выводящие его во внешнюю среду организма. Из-за дефекта структуры белка нарушается перемещение ионов натрия и хлора в клетках, что в итоге приводит к повышенной вязкости секрета экзокринных желез. Происходит увеличение всасывания ионов натрия и дефект секреции хлора, что ведёт к снижению или прекращению выделения жидкости в просвет бронхов. Из-за этого мокрота теряет свою жидкую часть, становится вязкой и густой. Впоследствии это ведёт к тому, что бронхи забиваются вязким секретом.

Таким образом, при муковисцидозе нарушаются функции желёз, которые вырабатывают пот, слизь, слёзы, слюну и пищеварительные соки. Через протоки этих желёз выделения выходят на поверхность тела или в полые органы, такие как кишечник или дыхательные пути. У пациентов с муковисцидозом в этих органах возникают серьёзные изменения, особенно они затрагивают бронхолёгочную систему — вязкий секрет закупоривает мелкие дыхательные пути.
Слизь, выделяемая в дыхательных путях, необходима для удаления из них пыли и бактерий, но при муковисцидозе выработка и структура бронхиального секрета нарушаются. В результате на стенках дыхательных путей развивается хроническое воспаление, которое впоследствии приводит к формированию бронхоэктазов (расширения и деструкции бронхов) и рубцов в лёгочной ткани.
При муковисцидозе страдает не только дренажная функция дыхательных путей, но и клеточный и гуморальный иммунитет. Это происходит из-за нарушений в работе клеток и затруднённого движения ресничек мерцательного эпителия бронхов.

Помимо этого, при муковисцидозе ферменты, вырабатываемые поджелудочной железой, не поступают в кишечник. Они начинают разрушать ткань поджелудочной железы, в результате чего в ней формируются рубцы и кисты. Также необратимо нарушается весь процесс пищеварения. Это приводит к недостаточности массы тела, отставанию в росте и весе, хронической диарее и выпадению прямой кишки.
При муковисцидозе у мужчин отмечается обструкция (непроходимость) семявыносящих протоков, отсюда и высокий процент мужского бесплодия при заболевании.
Классификация и стадии развития муковисцидоза
В зависимости от преобладающих симптомов выделяют три основные клинические формы заболевания: кишечную, бронхолёгочную и смешанную.
Кишечная форма муковисцидоза. Кистозный фиброз с кишечными проявлениями Е84.1 (код по МКБ-10)
Из-за неправильной секреции ферментов нарушается работа органов желудочно-кишечного тракта. Ухудшается расщепление жиров, белков и углеводов, а также их усвоение. Не вся пища переваривается, стул становится обильным, частым и зловонным.
Отмечается вздутие, повышенное газообразование и боли в различных областях живота. Даже при хорошем аппетите дети теряют вес, отстают в росте и развитии. Кишечная форма выявляется у 5—10 % больных муковисцидозом [7].
Бронхолёгочная форма муковисцидоза. Кистозный фиброз с лёгочными проявлениями Е84.0
Нарушения затрагивают всю бронхолёгочную систему. Вязкая мокрота скапливается в дыхательных путях и её тяжело откашлять. При инфицировании золотистым стафилококком, синегнойной и гемофильной палочками возникают тяжёлые бронхиты и пневмонии, для которых характерно осложнённое течение с развитием одышки, интоксикации и выраженной слабостью. Со временем развиваются следующие необратимые изменения:
- деформация грудной клетки;
- расширение дистальных отделов бронхов (бронхоэктазы);
- замещение лёгочной ткани соединительной рубцовой (пневмосклероз);
- «лёгочное сердце» — из-за повышенного давления в малом круге кровообращения происходит увеличение правых отделов сердца.
Бронхолёгочная форма характерна для 15—20 % случаев муковисцидоза [7].

Смешанная форма (лёгочно-кишечная)
Включает проявления лёгочной и кишечной форм. Это самый распространённый тип (65—75 % от всех случаев) и, как правило, самый тяжёлый [7].
Различают четыре стадии развития муковисцидоза:
1. Непостоянство симптомов, пациент жалуется на частый сухой кашель и небольшую одышку при физических нагрузках. Стадия может длиться до 10 лет.
2. Кашель с отделением мокроты, одышка появляется в покое и усиливается при физической нагрузке, фаланги пальцев и ногтевые пластины деформируются по типу «барабанных палочек» и «часовых стёкол». Стадия продолжается от 2 до 15 лет.
3. Бронхолёгочные симптомы прогрессируют, развиваются тяжёлые осложнения. У пациентов усиливается одышка и появляются признаки сердечной недостаточности: отёки на ногах, одышка при небольшой активности или в покое, быстрая утомляемость, непереносимость физических нагрузок. На УЗИ сердца видны признаки гипертрофии левых и правых отделов, а также снижение сердечной фракции выброса левого желудочка. На рентгенологических снимках можно обнаружить зоны пневмофиброза, бронхоэктазы и кисты. Формируется сердечная недостаточность по правожелудочковому типу, так называемое «лёгочное сердце». Длительность стадии от 3 до 5 лет.
4. Тяжёлая кардио-респираторная недостаточность. Из-за дисфункции левого желудочка нарушается работа сердца и лёгких, снижается фракция выброса левого желудочка, учащается сердцебиение, прогрессирует гипотензия. Стадия длится несколько месяцев и завершается смертью больного, вызванной развитием дыхательной недостаточности и остановкой сердечной деятельности.
Различают стадии обострения и ремиссии:
- лёгкое обострение — не чаще раза в год, в период ремиссии клинических проявлений нет, работоспособность пациентов не изменена;
- обострение средней тяжести — 2-3 раза в год, сохраняется кашель, периодически поднимается температура, страдает работоспособность;
- тяжёлые обострения – следуют одно за другим, ремиссии почти нет, масса тела больного снижается, больные становятся инвалидами.
Критерии обострения:
- увеличивается частота дыхания;
- повышается температура тела;
- вес снижается более чем на 1 кг;
- усиливается кашель;
- увеличивается количество мокроты;
- появляются хрипы;
- снижается ОФВ1 (объём форсированного выдоха за первую секунду манёвра) на 10 % и более в сравнении с измеренным за последние три месяца;
- снижается уровень сатурации кислорода на 10 % и более по сравнению с измерением за три последних месяца;
- ухудшается переносимость физических нагрузок.
Осложнения муковисцидоза
Для муковисцидоза характерны следующие бронхолёгочные осложнения:
- ателектаз — спадание части лёгкого;
- абсцессы в лёгких — развитие в них полости с гнойным содержимым.
Пациенты жалуются на выраженную одышку, отмечаются симптомы сильной интоксикации, повышается температура и выделяется большое количество зловонной гнойной мокроты.
Одним из тяжёлых осложнений является спонтанный пневмоторакс — скопление воздуха в плевральной полости. Нарушение проявляется резкой колющей болью в грудной клетке, усиливающейся при дыхании, кашле и движениях. Больной начинает дышать часто и поверхностно, отмечается сильная одышка и чувство нехватки воздуха, беспокоит кашель, появляется синюшность кожных покровов лица.

У больных старше 12 лет высока вероятность развития сахарного диабета I типа, может наблюдаться остеопороз, артриты и отёки нижних конечностей.
Постоянные или частые боли в животе могут указывать на панкреатит, эзофагит или язвенную болезнь желудка и двенадцатиперстной кишки.
Осложнять течение болезни может развитие фиброза печени различной степени выраженности, который чаще всего прогрессирует и в дальнейшем приводит к билиарному циррозу печени и портальной гипертензии. Муковисцидоз влияет на транспортировку ионов хлора, участвующих в перемещении желчи. При заболевании желчь по протокам не попадает в печень и её каналы, что ведёт к негативным последствиям — желчные протоки обезвоживаются, ткань печени замещаются соединительной тканью, происходит застой желчи (холестаз), развивается фиброз печени (цирроз)[11].
У больных муковисцидозом часто наблюдаются такие осложнения, как выпадение прямой кишки и кишечная непроходимость, связанные с вязкостью кала. Кишечная непроходимость вызвана дегидратацией кишечного содержимого, нарушением моторики кишечника и внешнесекреторной недостаточностью функции поджелудочной железы. При патологии пациенты жалуются на боль в животе, вздутие, запор, рвоту и задержку газов. Если больному вовремя не оказана специализированная медицинская помощь, то он может погибнуть от развившегося некроза и перитонита [12].
Диагностика муковисцидоза
Основной критерий постановки диагноза — увеличение концентрации ионов хлора в потовых железах более 60 ммоль/л вместе с одним или более из следующих критериев:
- хроническое заболевание дыхательных путей;
- экзокринная недостаточность поджелудочной железы;
- муковисцидоз близких – не далее двоюродных-родственников.
Эти критерии позволяют установить диагноз в 95 % случаев при недоступности генетического исследования мутации гена CFTR.
Характерные клинические проявления болезни:
- желудочно-кишечные нарушения;
- патологии бронхолёгочной системы и придаточных пазух носа;
- синдром потери солей;
- обструктивная азооспермия — сперматозоиды вырабатываются в нормальном количестве, но из-за нарушенной проходимости протоков не могут выйти наружу.
Скрининг на муковисцидоз проводится в роддоме всем новорождённым и направлен на выявление заболевания ещё до клинических проявлений. Для этого у ребёнка на 4-7 день жизни берётся кровь из пятки на фильтровальную бумагу и определяют уровень иммунореактивного трипсина (ИРТ). Если показатели ИРТ превышены (норма не более 65-70 нг/мл), то на 21-28 день анализ повторяют. Если показатели остаются повышенными, ребёнку проводится потовая проба и генетическое обследование.

Потовая проба — это один из методов диагностики муковисцидоза, с помощью которого определяется уровень хлоридов в секрете потовых желез. Анализ проводится троекратно. Положительным результатом для постановки диагноза считается показатель выше 60 ммоль/л.
Методика проведения потовой пробы: кожу правого бедра новорождённого протирают раствором натрия хлорида, дают высохнуть, после кладут салфетку, смоченную раствором пилокарпина для провоцирования гипергидроза и вторую салфетку, смоченную солёной водой. На салфетках закрепляются электроды, в течение 5 минут подаётся разряд 4 мА, после чего электроды снимаются, и кожа протирается. На простимулированном участке фиксируется кусочек фильтровальной бумаги. Выделяющийся пот собирается в течение 30 минут, после чего бумагу снимают и отсылают в лабораторию для определения концентрации электролитов хлора и натрия.
Обзорную рентгенограмму брюшной полости проводят для выявления вздутых петель кишечника и признаков кишечной непроходимости. При осмотре может быть обнаружено увеличение печени (гепатомегалия). Также у пациентов выявляют сахарный диабет I типа, который сочетается с респираторными симптомами: одышкой, частым кашлем с трудноотделяемой вязкой мокротой и панкреатитом, проявляющимся опоясывающими болями в области живота и частым стулом непереваренной пищей.

Обследование в несколько этапов необходимо, чтобы исключить ложноположительные результаты и подтвердить или опровергнуть диагноз [6].
Молекулярно-генетический анализ позволяет диагностировать муковисцидоз приблизительно в 90 % случаев. Для его проведения нужна кровь из вены. Генетический метод обнаруживает в хромосомах пациента «дефектный» ген с мутацией F508del. При тестировании на муковисцидоз невозможно проверить все мутации, так как их более 1900, а большинство лабораторий выявляет только 20-30 самых частых вариантов.
Если в хромосомах не обнаруживается ни одной из 10-20 наиболее распространённых мутаций, то диагноз «муковисцидоз» маловероятен. В России генетические анализы недостаточно распространены, и не во всех регионах есть возможность такого тестирования [8].
Лечение муковисцидоза
Муковисцидоз носит наследственный характер, поэтому его лечение большей частью симптоматическое, оно направлено на восстановление функций дыхательного и желудочно-кишечного тракта и проводится на протяжении всей жизни пациента. Больным муковисцидозом важно соблюдать диету, выполнять специальный комплекс упражнений и принимать препараты, предотвращающие развитие инфекций.
Основной целью лечения при муковисцидозе является удаление из дыхательных путей вязкой мокроты. Для этого применяют различные методики дренирования бронхиального дерева, например кинезотерапию — одну из форм лечебной физкультуры, способствующую очищению бронхов от вязкой и агрессивной слизи.
Приёмы кинезотерапии:
- Углублённые вдох и выдох, позволяющие слизи легче отделяться от стенок бронхов.
- Дыхание с сопротивлением губами.
- Мобилизация — упражнения для повышения эластичности грудной клетки, позвоночника и мышечного баланса тела.
Физиотерапевтическое лечение младенцев и детей раннего возраста включает как активную, так и пассивную техники.
Пассивная техника:
• принятие телом ребёнка определённого положения на короткое время, затем его изменение;
• сопровождение и стимуляция дыхательных движений при помощи рук (контакт-дыхание) — техника, при которой рука физиотерапевта ведёт и стимулирует дыхательные движения;
• ручная вибрация на выдохе — для поддержки выдоха и помощи высвобождению мокроты потряхивается грудную клетку рукой;
• потряхивание — ритмические движения, выполняемые в одной из частей тела для возникновения реакции в грудной клетке;
• терапевтические положения тела — расположение грудной клетки в состоянии, при котором растягивается лёгочная ткань и расширяются дыхательные пути.

Активная техника:
• влияние на дыхание, например с помощью акустической стимуляции — поощряя ребёнка повторять громкие крики, которые он издаёт;
• упражнения для развития эластичности грудной клетки, позвоночника и мышечного баланса тела;
• упражнения, повышающие ловкость, выносливость и формирующие наслаждение от мышечной активности.
Важную роль при лечении играют аппаратные методы. Широкое распространение получил аппарат «виброжилет», состоящий из надувного жилета и блока генератора пневмоимпульсов, который создаёт давление на грудную клетку. Такие колебания воспроизводят эффект кашля и помогают очистить бронхи от вязкой мокроты. Аппаратные методы применяют вместе с медикаментозной терапией [13].

При муковисцидозе важно соблюдать диету — питание должно быть высококалорийным, с повышенным содержанием белков и жиров. Пищу принимают часто и небольшими порциями, дополнительно подсаливая. Из рациона исключается еда, которая тяжела для переваривания: грубая клетчатка, навары из супов, жареные блюда, шоколад, торты, пирожные. Суточный калораж должен превышать возрастную норму на 20—40 %, главным образом за счёт увеличенного количества белка (до 6 г на 1 кг массы тела в сутки). Из-за нарушенной функции поджелудочной железы у больных не усваиваются жиры и жирорастворимые витамины А, Д, Е и К, поэтому необходим их дополнительный приём.
Для облегчения отхождения мокроты применяют следующие препараты:
- Амброксол в таблетках по 30 мг. В период ремиссии лекарство принимают один раз в сутки, во время обострения заболевания — до шести раз.
- Дорназа альфа — генно-инженерный вариант природного фермента человека, который помогает расщеплять гнойную мокроту. Применяется с помощью небулайзера по 2,5 мг один раз в сутки.

Для ингаляционной терапии используют бронхолитики:
- «Беродуал» — на одну процедуру в небулайзере нужно 20 капель беродуала + 3,0 мл физраствора. Ингаляции делают два раза в день утром и вечером в течение 7-10 дней.
- «Вентолин» — препарат заливают в небулайзер в неразбавленном виде, стандартная доза 2,5 мл, в сутки разрешается делать не больше четырёх ингаляций.
- «Пульмикорт» — на одну процедуру нужно 1 мл препарата + 1 мл. физраствора, дважды в день утром и вечером в течение пяти дней.
Следует иметь в виду, что у трети больных бронхолитики оказывают положительный ответ, но у большей части может возникнуть коллапс мышц дыхательных путей и резкое снижение объёма выдыхаемого воздуха. В связи с этим больные, получающие такую терапию, должны находиться под наблюдением врача.
Антибактериальные препараты назначаются всем пациентам с лёгочными симптомами, при обострениях заболевания или выявлении возбудителей респираторных инфекций для подавления их роста.
Для лечения применяют эффективные комбинации антисинегнойных антибиотиков:
Ингаляционные антибактериальные средства: «Брамитоб», «Тобрамицин», «Тоби подхалер», «Колистин».
Пероральные антибиотики: ципрофлоксацин, кларитромицин.
Парентеральные антибиотики:
- цефалоспорины + аминогликозиды;
- антисинегнойные пенициллины + аминогликозиды;
- карбапенемы + аминогликозиды;
- фторхинолоны + аминогликозиды;
- цефалоспорины + фторхинолоны;
- антисинегнойные пенициллины + фторхинолоны;
- карбапенемы + фторхинолоны;
- цефалоспорины + карбапенемы + аминогликозиды [10].
Доза вводимого антибактериального препарата должна превышать среднюю терапевтическую в полтора раза. Курс антибактериальной терапии может продолжаться до 3-4 недель,в случае хронизации инфекционного процесса — до 12 недель.
Для улучшения функции пищеварения используют ферментные препараты, которые содержат липазу, протеазу и амилазу, облегчающие переваривание жиров, углеводов и белков, а также способствующие их лучшему всасыванию в тонком кишечнике. Подбор доз панкреатических ферментов проводится индивидуально исходя из клинических проявлений болезни. Применяются препараты: «Панкреатин», «Мезим», «Креон». Ферментные препараты назначаются в больших дозах, чем больным с другими заболеваниями желудочно-кишечного тракта — до 30 капсул «Креона» в сутки.
Лечение муковисцидоза пожизненное. Выбор схемы терапии зависит от вида бактерий, обнаруженных в бронхиальном секрете дыхательных путей, а также от преобладающих клинических форм заболевания.
«В последние годы для лечения муковисцидоза начали применять таргетную генетическую терапию, которая направлена на исправление дефекта в самой распространённой мутации F508del [14]. Препарат продаётся в США под маркой «Trikafta». В августе 2020 года лекарство было одобрено Европейской комиссией для применения в ЕС под наименованием «Kaftrio»». — прим. ред. «ПроБолезни».
В России оригинальные препараты для лечения муковисцидоза заменяются дженерикам отечественного производства. Дженерики сходны с оригинальными препаратами по составу действующего вещества. Многие пациенты жалуются на их плохое качество и появление побочных эффектов. В последнее время пациентами и родителями больных муковисцидозом проводятся кампании, цель которых — вернуть государственное обеспечение качественными иностранными лекарствами.
Назвать побочные эффекты дженериков на данный момент сложно, так как отсутствуют их сравнительные исследования с оригинальными препаратами.
В рамках 44-ого ФЗ закупки лекарственных препаратов идут по международному непатентованному названию. Перечень лекарственных препаратов утверждается правительством РФ. Врачам за назначение препаратов под торговыми названиями или купленными родителями и благотворительными фондами грозит ответственность. В рецептах, выписанных врачом, должны быть указаны только международные названия, а не торговые. За нарушение этого правила врачей ждет штраф.
Хотя есть одно условие: при наличии медицинских показаний (аллергия, индивидуальная непереносимость дженерика по жизненным показаниям) по решению врачебной комиссии пациент может получить лекарство под торговым названием. Поэтому возможность получить оригинальный препарат — индивидуальная непереносимость дженерика (которую нужно доказать) и решение врачебной комиссии.
Муковисцидоз — наиболее частое показание для трансплантации лёгких. Основные показания для этой операции: прогрессирующее снижение веса, ухудшение лёгочной функции, частые обострения, потребность в кислородотерапии. Противопоказания: аспергиллома (опухолевоподобное неинвазивное новообразование шаровидной формы, состоящее в основном из клеток мицелия микроскопического плесневого гриба), курение, психосоциальные факторы. Сахарный диабет и лечение кортикостероидами не являются препятствием для трансплантации.
Прогноз. Профилактика
Муковисцидоз — это неизлечимое хроническое заболевание, но при своевременной диагностике и адекватной постоянной терапии многие пациенты доживают до взрослого возраста.
Для больных муковисцидозом возможна полноценная жизнь, но только при условии постоянного получения необходимых препаратов. Основными причинами смертности этих пациентов являются бронхолёгочные осложнения: развитие инфекции в нижних дыхательных путях, бронхоэктазы и хроническая обструктивная болезнь легких (ХОБЛ).
Доля пациентов старше 18 лет за последнее 10 лет увеличилась, выживаемость составила 31,8 % [3]. Увеличение продолжительности жизни обусловлено ранним выявлением заболевания, оптимизацией работы центров муковисцидоза и применением эффективных медикаментозных препаратов, в частности дорназы альфа («Пульмозим»), некоторых ингаляционных и системных антибиотиков и макролидов [9]. Дорназа альфа способствует снижению вязкости мокроты и обладает отхаркивающим действием. Использование небулайзера помогает быстро доставить ингаляционные антибиотики в поражённую область.
Главным способом профилактики является дородовая и неонатальная диагностика. Семейным парам, планирующим рождение ребенка, рекомендуется пройти ДНК-диагностику на носительство мутаций, приводящих к муковисцидозу, с последующей консультацией врача-генетика.
Пренатальная ДНК-диагностика выполняется при заборе околоплодных вод в ранний (13-14 недель) и поздний (16-20 недель беременности) сроки. Обследование проводят для семей, в которых выявлено носительство мутации гена CFTR и имеющих ребёнка больного муковисцидозом.
- ИНВИТРО
- Библиотека
- Справочник заболеваний
- Муковисцидоз…
Муковисцидоз (кистозный фиброз)
Муковисцидоз: причины появления, симптомы, диагностика и способы лечения.
Определение
Муковисцидоз – это мультисистемное, наследственное, опасное для жизни заболевание, характеризующееся поражением экзокринных желез, а также жизненно важных органов и систем: дыхательных путей, желудочно-кишечного тракта, поджелудочной железы, печени, слюнных и потовых желез, репродуктивной системы.
В России частота заболевания составляет 1 на 9000 новорожденных. В прошлом (из-за высокой ранней смертности) муковисцидозом болели в основном дети. Современный уровень диагностики и лечебных мероприятий позволяет значительно продлить жизнь таких больных – до 35-50 лет. В Москве медиана выживаемости достигает 39,5 лет.
Причины появления муковисцидоза
Причиной патологических изменений при муковисцидозе является мутация гена, находящегося в длинном плече 7-й хромосомы и передающегося по аутосомно-рецессивному типу при наследовании двух мутантных генов родителей. Ген назван муковисцидозным трансмембранным регулятором (МВТР). На сегодняшний день выделено более 2000 мутаций этого гена. Он регулирует транспорт электролитов (главным образом хлора) через мембраны эпителиальных клеток, выстилающих выводные протоки экзокринных желез. Мутация приводит к нарушению структуры и функции синтезируемого белка, в результате чего секрет, выделяемый этими железами, становится чрезмерно густым и вязким.
Название заболевания происходит от латинских слов mucus – «слизь» и viscidus – «вязкий».
В патогенезе данного генетического заболевания выделяют следующие патологические звенья:
- нарушение функции экзокринных желез;
- нарушение электролитного обмена;
- поражение соединительной ткани, что часто связано с формированием иммунных комплексов к синегнойной палочке.
Классификация заболевания
В современной классификации выделяют:
- Классический муковисцидоз:
- с панкреатической недостаточностью (смешанная или легочно-кишечная форма заболевания);
- с ненарушенной функцией поджелудочной железы (преимущественно легочная форма заболевания);
- неуточненный – неопределенный диагноз при положительном неонатальном скрининге на муковисцидоз.
- Атипичный муковисцидоз протекает в одной из форм: хронический панкреатит, изолированная обструктивная азооспермия, диссеминированные бронхоэктазы, аллергический бронхолегочный аспергиллез, диффузный панбронхиолит, склерозирующий холангит.
Симптомы муковисцидоза
Тяжесть протекания муковисцидоза напрямую зависит от возраста, когда стали проявляться первые симптомы, — чем младше ребенок, тем тяжелее течение заболевания и неблагоприятнее прогноз. У большинства детей отмечаются часто рецидивирующие респираторные инфекции с явлениями бронхита. Достаточно рано появляется кашель с выделением гнойной мокроты, тошнотой, рвотой и нарушением сна. Также может отмечаться свистящее дыхание, одышка, задержка прибавки веса, частый, с примесью жира и зловонным запахом стул.
Кожные покровы имеют «соленый» привкус в связи с отложением кристаллов соли на коже ребенка.
Симптомы в разные возрастные периоды имеют свои особенности:
- у новорожденных к основным симптомам относят рвоту с примесью желчи, отсутствие стула, вздутие живота, продолжительную обструктивную желтуху, кишечную (мекониевую) непроходимость, выраженный сосудистый рисунок на коже живота, интоксикацию, полигиповитаминоз, быстрое сморщивание кожи пальцев в воде, бронхообструктивный синдром, гипонатриемию и гипокалиемию, анемию, выбухание родничка и паралич лицевого нерва, отставание в физическом развитии, недостаточность ферментативной активности желудочно-кишечного тракта (особенно ярко проявляется после перевода ребенка на искусственное вскармливание или прикорм), быстро развивающуюся гипотрофию;
- у детей дошкольного возраста преобладают бронхообструктивный синдром, рецидивирующие синуситы, полипы носа, непереносимость жары, выпадение прямой кишки, панкреатит, гепатомегалия и др.;
- у детей школьного возраста в мокроте выделяют синегнойную палочку, бронхоэктазы, симптом «барабанных палочек», нарушение углеводного обмена, наблюдаются полиурия, полидипсия, снижение массы тела, цирроз печени, гипертензия, спленомегалия, асцит, варикозное расширение вен пищевода, задержка физического развития.
 У заболевших в раннем детстве и доживших до взрослого возраста отмечаются следующие симптомы: постоянно рецидивирующие инфекционно-воспалительные процессы в легких, изменения бронхиальной стенки, бронхоэктазы, пневмофиброз, буллезная и обструктивная эмфизема, цирротические изменения, сахарный диабет, синуситы и др.
У заболевших в раннем детстве и доживших до взрослого возраста отмечаются следующие симптомы: постоянно рецидивирующие инфекционно-воспалительные процессы в легких, изменения бронхиальной стенки, бронхоэктазы, пневмофиброз, буллезная и обструктивная эмфизема, цирротические изменения, сахарный диабет, синуситы и др.
 Альвеолярный мешок здорового человека (слева) и альвеолярный мешок пациента с муковисцидозом (справа)
Альвеолярный мешок здорового человека (слева) и альвеолярный мешок пациента с муковисцидозом (справа)
Взрослые с поздней манифестацией и атипичной формой заболевания имеют скрытые формы болезни, которые чаще всего случайно проявляются в виде рецидивирующего бронхита, синусита, хронической обструктивной болезни легких, цирроза печени, бесплодия.
Диагностика муковисцидоза
Диагностика муковисцидоза основана на данных лабораторных и инструментальных исследований, клинической картине, физикальном обследовании и тестах функциональной диагностики.
Скрининг новорожденных на муковисцидоз направлен на доклиническое выявление патологии и основан на определении иммунореактивного трипсина в сухом пятне капиллярной крови.
Образец крови берется у доношенного ребенка из пятки на 4-й день жизни через 3 часа после кормления и на 7-й день – у недоношенного.
К специфическим лабораторным критериям муковисцидоза относятся:
- потовый тест — определение электролитов пота (показывает концентрацию ионов натрия и/или хлора);
- исследование назальных потенциалов;
- исследование панкреатической эластазы кала;
Муковисцидоз, CFTR ч.м.
Исследование частых мутаций в гене CFTR.
Тип наследования.
Аутосомно-рецессивный.
Гены, ответственные за развитие заболевания.
CFTR -трансме…
Основные наследственные заболевания
Определение носительства частых мутаций в генах, ответственных за развитие наиболее частых аутосомно-рецессивных заболеваний: муковисцидоз, несиндромальная ней�…
Основными инструментальными методами диагностики муковисцидоза являются:
- рентгенография;
- исследование функции внешнего дыхания;
Эхокардиография
Исследование, позволяющее оценить функциональные и органические изменения сердца, его сократимость, а также состояние клапанного аппарата.
Если ребенку поставлен диагноз «муковисцидоз», то генетический анализ потребуется всем членам семьи. Это важно не только для подтверждения диагноза, но и для дородовой диагностики при последующих беременностях.
Дифференциальная диагностика муковисцидоза проводится с туберкулезом, мекониальным илеусом, желтухой новорожденных, гемолитической анемией, выпадением прямой кишки, рецидивирующими бронхитом, синуситом, бронхиальной астмой, бронхоэктазами и другими заболеваниями.
К каким врачам обращаться
Если у ребенка наблюдаются какие-то из перечисленных симптомов, следует безотлагательно сообщить об этом
врачу-педиатру
. При подозрении на муковисцидоз участковый педиатр дает направление в Российский или региональный центр по борьбе с муковисцидозом. Лечение муковисцидоза представляет собой трудную задачу, которая требует консультаций
пульмонолога
,
гастроэнтеролога
и других специалистов.
Лечение муковисцидоза
Терапия муковисцидоза носит комплексный характер и направлена на разжижение и удаление вязкой мокроты из бронхов, борьбу с инфекционными заболеваниями легких, замещение недостающих ферментов поджелудочной железы, коррекцию поливитаминной недостаточности, разжижение желчи.
Важное место в терапии бронхо-легочной формы муковисцидоза занимает кинезотерапия (специальный комплекс упражнений и дыхательной гимнастики, направленных на удаление мокроты). Занятия должны быть ежедневными и пожизненными. Необходимо обеспечить физическое развитие ребенка согласно возрастным нормам, максимально высокое качество жизни пациента, профилактику осложнений.
В зависимости от тяжести состояния лечение может проводиться в специализированном отделении больницы, в дневных стационарах или на дому. Такое лечение включает:
- проведение активной муколитической терапии;
- назначение бронхолитической терапии (ингаляционных бронходилататоров, в некоторых случаях кортикостероидов);
- антибактериальную терапию, включая ингаляционную;
- противогрибковую терапию (по показаниям);
- лечение гепатотропными препаратами;
- высококалорийную диету (иногда требуется дополнительное энтеральное зондовое питание).
Хирургические методы лечения муковисцидоза:
- лапаротомия с резекцией части кишки (в период новорожденности при мекониальном илеусе);
- полипэктомия (при полипозе носа);
- постановка порт-системы (венозного катетера, имплантируемого под кожу) — при длительных курсах внутривенной антибактериальной терапии;
- спленэктомия, шунтирующие и склерозирующие манипуляции (при портальной гипертензии);
- плеврэктомия, плевродез (при повторных пневмотораксах).
При тяжелом течении заболевания необходимо пребывание пациента в условиях стационара с длительной кислородотерапией и практически постоянный прием антибактериальных препаратов.
При соблюдении этих условий терминальная фаза болезни может продолжаться несколько лет. В этой фазе больные планируются на пересадку легких.
Очень важную роль в лечении детей с муковисцидозом играют родители. На их плечи ложится большая ответственность по уходу за больным ребенком, поскольку терапия этого заболевания пожизненная и требует скрупулезного выполнения всех рекомендаций врача. Только родители, постоянно находясь с ребенком, могут оценить изменение в состоянии малыша и вовремя обратиться за медицинской помощью.
Осложнения
Основной причиной осложнений и летальности являются патологии дыхательных путей. К неблагоприятным и тяжелым последствиям муковисцидоза относятся:
- абсцессы;
- ателектазы;
- пневмоторакс, пиопневмоторакс;
- гипоксемия;
- гиперкапния;
- легочное сердце;
- желудочное и легочное кровотечение;
- отечный синдром;
- сахарный диабет;
- печеночная энцефалопатия;
- амилоидоз почек;
- острая и хроническая почечная недостаточность;
- фиброз печени, гепатоцеллюлярная недостаточность, желчнокаменная болезнь;
- неукротимая рвота;
- диарея, приводящая к обезвоживанию и выраженным электролитным нарушениям и др.
Рецидивирующие респираторные заболевания в виде бронхитов, бронхиолитов, пневмонии приводят к еще большему увеличению вязкости мокроты, обструкции дыхательных путей, инфекциям и частым воспалениям.
Повышенная вязкость секрета желез внешней секреции опасна хроническим воспалительным процессом в легких, экзокринной недостаточностью поджелудочной железы, гепатобилиарной патологией и аномально высоким содержанием электролитов в поте.
Вязкий кишечный секрет может привести к мекониальной непроходимости у новорожденных (мекониальному илеусу) и иногда к синдрому мекониевой пробки. У детей старшего возраста и взрослых также могут быть симптомы хронического запора и непроходимости кишечника.
Другие проблемы со стороны желудочно-кишечного тракта включают инвагинацию кишечника, странгуляционную кишечную непроходимость, ректальный пролапс, периаппендикулярный абсцесс, панкреатит, повышенный риск развития рака гепатобилиарного и желудочно-кишечного трактов (включая рак поджелудочной железы), гастроэзофагеальный рефлюкс, эзофагит, болезнь Крона и целиакию.
Бесплодие наблюдается у 98% мужчин из-за недоразвития семявыносящих протоков или других форм обструктивной азооспермии. У женщин фертильность несколько снижается из-за вязкого цервикального секрета, хотя многие женщины вынашивают беременность полностью. Исходы беременности для матери и новорожденного связаны со здоровьем матери.
Другие осложнения включают остеопению/остеопороз, депрессию и беспокойство, хроническую боль, обструктивное апное во сне, почечные камни, диализозависимую хроническую почечную недостаточность, железодефицитную анемию и эпизодические артралгии/артриты.
Течение заболевания во многом определяется степенью поражения легких. Ухудшение неизбежно, что приводит к истощению и, в конечном итоге, к смерти, как правило, из-за сочетания дыхательной недостаточности и легочного сердца. Прогрессирование легочной и сердечной недостаточности является наиболее частой причиной смерти пациентов (95%). Среди других причин выделяют осложнения при трансплантации органов (12%), заболевания печени и печеночную недостаточность (2,3%), травмы (2,1%), суицид (0,8%), другие (1,3%).
Профилактика муковисцидоза
Если в семье есть случаи муковисцидоза, то при планировании беременности обязательно следует обратиться к медицинскому генетику. В настоящее время стала возможной дородовая диагностика муковисцидоза у плода. Именно поэтому при возникновении каждой новой беременности необходимо сразу же (не позднее 8 недели беременности) обратиться в центр дородовой диагностики.
В конце ХХ века начали создаваться Российские и региональные центры по борьбе с муковисцидозом. Основу терапевтической помощи больным составляет грамотно подобранная пожизненная лекарственная терапия, регулярные профилактические осмотры и стационарное лечение в период обострений.
Источники:
- Клинические рекомендации «Кистозный фиброз (муковисцидоз)». Разраб.: Союз педиатров России, Ассоциация медицинских генетиков, Российское респираторное общество. – 2019. – 89 с.
- Баранов А.А., Намазова-Баранова Л.С., Симонова О.И., Каширская Н.Ю. и соавт. Современные представления о диагностике и лечении детей с муковисцидозом // Педиатрическая фармакология. – 2015; 12 (5): 589–604.
- Aurora P, Gustafsson P, Bush A, et al: Multiple breath inert gas washout as a measure of ventilation distribution in children with cystic fibrosis. Thorax 59:1068–1073, 2004.
ВАЖНО!
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Информация проверена экспертом

Лишова Екатерина Александровна
Высшее медицинское образование, опыт работы — 19 лет
Поделитесь этой статьей сейчас
Рекомендации
-
3751
09 Февраля
-
3745
09 Февраля
-
3769
08 Февраля
Похожие статьи
Гепатит С
Гепатит С: причины появления, классификация, симптомы, диагностика и способы лечения.
Миеломная болезнь
Миеломная болезнь, или множественная миелома: причины появления, симптомы, диагностика и способы лечения.
Полипы желудка
Полипы желудка: причины появления, симптомы, диагностика и способы лечения.
Энтеробиоз
Энтеробиоз: причины появления, симптомы, диагностика и способы лечения.
 |
| У врачей МВ традиционно ассоциируется прежде всего с патологией легких, однако он представляет собой системное заболевание с поражением желудочно-кишечного тракта и других жизненно важных органов |
В медицинской практике МВ традиционно ассоциируется прежде всего с патологией легких, однако он представляет собой системное заболевание с поражением желудочно-кишечного тракта, вторично — сердца, других жизненно важных органов. И хотя прогноз остается неблагоприятным и не все больные переживают 25-летний возрастной рубеж, МВ можно рассматривать как многодисциплинарную клиническую проблему, прежде всего на стыке педиатрии и терапии
Муковисцидоз (МВ) — генетическое аутосомно-рецессивное (а/р) моногенное заболевание, обусловленное мутацией гена трансмембранного регулятора МВ (МВТР). Характеризуется нарушением секреции экзокринных желез жизненно важных органов с поражением прежде всего дыхательного и желудочно-кишечного тракта, тяжелым течением и неблагоприятным прогнозом [1, 2, 3]. Впервые выделено из группы целиакий в 1936 г. венским педиатром Гвидо Фанкони [9].
Первоначальная мутация возникла, вероятнее всего, где-то на границе современных Голландии и Германии. Ген обнаруживается, по европейским данным, с частотой 1 случай на 1500 человек, уступая по распространенности только синдрому трисомии [12]. Постепенно распространяется на восток, где заболеваемость явно нарастает одновременно как результат улучшения диагностических мер и как истинный показатель. Вероятность рождения больного ребенка, по европейским данным, составляет 1:2000 — 1:2500 живорожденных [13]. Число диагностированных больных в развитых странах равно 7-8 на 100 тыс. населения, из них количество больных старше 18 лет составляет 20%, а в США даже 32% [8]. В России большая часть этих больных не выявляется или выявляется поздно, нередко в запущенной стадии болезни, и поэтому распространенность МВ по нашей статистике не превышает 1:100 000, по расчетам же медико-генетического центра РАМН она должна быть 1:12 000 новорожденных [4].
После первых описаний МВ считался фатальным заболеванием, так как большинство детей не переживало пятилетний рубеж. До 80-х годов 80% больных не достигало возраста 20 лет [13]. Но и в современных условиях существует большой разброс ожидаемой продолжительности жизни больных, что обусловлено уровнем развития общества, осознанием актуальности проблемы, степенью организации специализированных центров. В Великобритании, США, Австралии новорожденным с МВ гарантируется 40 лет жизни, в других развитых странах — в среднем 31, в Латинской Америке — 10 лет (95% больных МВ здесь не выявляется), в России, по результатам работы центра МВ на базе республиканской детской больницы (Москва), — 16 лет. Лечение МВ требует больших финансовых вложений. Так, в США они составляют 15 тыс. долл. в год, в Великобритании — 11 тыс. ф. ст., в Российском центре МВ — 10 500 долл. в год [2].
Таким образом, МВ является важной медико-социальной проблемой, что обусловлено не только большими моральными, физическими и материальными затратами семьи, органов практического здравоохранения и общества в целом на диагностику, лечение и социальную адаптацию больных, но и достижениями научно-практической медицины, позволившими увеличить продолжительность жизни больных.
Ген МВ локализован на 7-й хромосоме (7q31). Передается а/р с различной степенью экспрессивности признаков. В одном гене может быть множество мутаций, каждая из которых характерна для определенной популяции или региона. Основная мутация дельта-F508, т. е. замена аминокислоты фенилаланина в 508-й позиции, встречается в России в 56% случаев, а в Москве — в 41%. Частота других мутаций — W1282X, N1303K и т. д. — 3-5% [4, 5, 11]. Тяжелое течение МВ свойственно гомозиготам по дельта-F508, а также при мутациях W1282X, G542X, N1303K как в гомозиготном состоянии, так и в сочетании с дельта-F508. Относительно легкое течение МВ наблюдается у детей с мутациями R334W, S1196X (даже в сочетании с дельта-F508). Но предсказать течение и прогноз болезни даже при одной и той же мутации сложно. Иногда при ранней диагностике МВ и адекватном лечении быстро развиваются необратимые изменения жизненно важных органов. В то же время у некоторых пациентов даже при поздней диагностике и соответственно позднем лечении наблюдалось благоприятное течение МВ, позволявшее им достичь взрослого, а иногда и зрелого возраста. Такой клинический полиморфизм можно объяснить генетическими особенностями самого ребенка, а также характером жизни, иммунными реакциями и т. д.
Патогенез МВ связан с дефектом синтеза белка, выполняющего роль хлоридного канала, участвующего в водно-электролитном обмене эпителиальных клеток дыхательных путей, желудочно-кишечного тракта, поджелудочной железы, печени, репродуктивной системы. В 70% мутаций в европейской популяции наблюдается потеря аминокислоты фенилаланина в белке. В связи с неспособностью дефектного белка адекватно выполнять работу хлоридного канала внутри клетки накапливаются ионы Cl–. Изменяется электрический потенциал в просвете выводных протоков, и внутрь клетки устремляется ион натрия. Последний в свою очередь выполняет роль насоса, что обусловливает усиленное всасывание воды из околоклеточного пространства [7]. В итоге сгущается секрет большинства желез внешней секреции, затрудняется его эвакуация, в органах возникают вторичные изменения, наиболее серьезные — в бронхолегочной системе. В стенках бронхиального дерева развивается хроническое воспаление различной выраженности, разрушается соединительнотканный каркас, формируются бронхиоло- и бронхоэктазы. В условиях постоянной обструкции вязкой мокротой и прогрессирующей деструкции легочной паренхимы бронхоэктазы становятся распространенными, нарастает гипоксия, развивается легочная гипертензия и так называемое «легочное сердце». Патогенез МВ включает в себя и другие механизмы. В частности, наблюдается уменьшение продукции IgА, снижение противовирусного иммунитета, образование интерферона, фагоцитарная активность лейкоцитов, особенно их микробицидное действие [1]. Альвеолярный макрофаг — основной фагоцит легких [7] — главный источник интерлейкина 8 (ИЛ8), ведущего хемоатрактанта для нейтрофилов. У больных МВ повышается концентрация ИЛ8 в мокроте и бронхоальвеолярной жидкости, положительно коррелирующая с тяжестью бронхолегочного процесса [10, 15]. Одновременно в большом количестве при МВ обнаруживают другие хемоатрактанты (С5а, лейкотриен В4), цитокины (ИЛ1, ИЛ6, альфа-фактор некроза опухоли), эластаза, играющие важную роль в формировании хронического воспаления [15]. Множество нейтрофилов, привлеченных в дыхательные пути, увеличивает количество гнойной мокроты. Разрушающиеся нейтрофилы освобождают ДНК, также увеличивающую вязкость мокроты. Эндо- и панбронхиту предшествует вирусное поражение носоглотки, гортани и бронхов, приводящее к гибели клеток мерцательного эпителия и открывающее путь для микробной флоры (тем более на фоне дефицита JgА). Как повреждающие агенты заявляют о себе бактерии, особенно синегнойная палочка, и эндогенные протеазы, прежде всего эластаза. Защитными факторами против последней являются альфа-один-антитрипсин и секреторный ингибитор лейкопротеаз, но у больных МВ они подавляются огромным количеством нейтрофильной протеазы. Это позволяет протеазам непосредственно разрушать эпителий и каркас бронхиального дерева, еще более изменяя мукоцилиарный клиренс. На ранних стадиях МВ в микробном пейзаже бронхов преобладает стафилококк, позднее — синегнойная палочка, что объясняется способностью патологического белка МВ изменять условия формирования и количественный состав «сахаров» на поверхности эпителиальной клетки дыхательных путей. Это облегчает адгезию микробов, особенно синегнойной палочки [11, 15, 16].
Бронхолегочные изменения преобладают в клинической картине и определяют прогноз у 95% пациентов [13, 16]. У врачей МВ традиционно ассоциируется прежде всего с патологией легких, однако он представляет собой системное заболевание с поражением желудочно-кишечного тракта, вторично — сердца, других жизненно важных органов. И хотя прогноз остается неблагоприятным и не все больные переживают 25-летний возрастной рубеж, МВ можно рассматривать как многодисциплинарную клиническую проблему, прежде всего на стыке педиатрии и терапии.
Пациенты с муковисцидозом и члены их семей должны наблюдаться в специализированных центрах. Но в нашей стране таких центров немного, а с учетом расстояний и отсутствия необходимых средств на дорожные расходы они и вовсе становятся малодоступными для пациентов. Поэтому принципы ведения больных с МВ должны быть знакомы врачам широкой лечебной сети.
- Клиническая картина
Выраженный полиморфизм МВ определяет разные варианты его течения. В 8-20% гомозиготных случаев МВ манифестирует с рождения мекониальным илеусом с возможным исходом в мекониальный перитонит. В менее трагичных ситуациях стул обильный, с сильным отвратительным запахом, «жирный». У 1/3 больных наблюдается выпадение прямой кишки, но при назначении адекватной дозы современных пищеварительных ферментов это осложнение спонтанно проходит через 1,5-2 мес. Следует напомнить, что мекониальный илеус, как правило, указывает на наличие МВ у новорожденного (дифференциальная диагностика — ДД — проводится прежде всего [14] с синдромом семейной хлоридной диареи). У больных школьного возраста первыми симптомами могут быть «кишечные колики». При обследовании таких детей в брюшной полости пальпируются «плотные образования» (каловые массы, смешанные с густой плотной слизью), которые вызывают вздутие живота, повторные рвоты, запоры. Дистальная кишечная обструкция или эквивалент мекониального илеуса в его острой, подострой или хронической форме могут наблюдаться у подростков, юношей и даже взрослых [11, 14, 15], вызывая необходимость в проведении ДД с синдромами висцерального обкрадывания, верхней брыжеечной артерии, полипозов, врожденной и приобретенной интестинальной псевдообструкции.
После назначения ферментов кишечная симптоматика отодвигается на второй план, уступая место легочной. Острый дебют бронхолегочной патологии наблюдается нечасто. Обычно постепенно развивается хронический (в 1/3 случаев — обструктивный) бронхит. При рождении дыхательные пути интактны, но уже в периоде новорожденности и грудном возрасте возникает кашель, приступы удушья, одышка, иногда рвота, рецидивирующие бронхопневмонии. Затем формируются эмфизема, нередко — ателектазы, быстро появляются бронхоэктазы. Периодически возникает мучительный сильный кашель, особенно по ночам. Мокрота вязкая, иногда гнойная. Может быть симптоматика бронхиальной астмы. Вначале аускультативная картина вне обострений остается неизменной, но при тщательном обследовании обнаруживается небольшая одышка, увеличение объема грудной клетки в основном за счет переднезаднего размера, незначительное, но постоянное снижение экскурсии грудной клетки. Как следствие перегрузки малого круга развивается «легочное сердце». Хроническая гипоксия приводит к деформации концевых фаланг пальцев по типу «барабанных палочек» и ногтей — по типу «часовых стекол».
Поражение поджелудочной железы обусловливает синдром мальабсорбции с дистрофией, обильным жирным стулом. На поздних стадиях развивается сахарный диабет и — у 13% больных смешанной и кишечной формой МВ — цирроз печени. Цирроз печени типичен для мутаций W1282X, дельта-F508 и Х1303К. У 5-10% больных выявляют билиарный цирроз с портальной гипертензией. В целом же клинически, лабораторно, инструментально и морфологически изменения печени обнаруживают у 86% больных [2, 15, 16].
Так как поражаются все органы, содержащие слизеобразующие железы, типичны колитический синдром, хронический холецистит, синуиты.
- Диагноз и принципы наблюдения [1, 12, 16]
1. Общие симптомы: дистрофия, отставание в физическом развитии, рецидивирующие хронические заболевания органов дыхания, полипы носа, упорно текущий хронический гайморит, хронический бронхит, рецидивирующий панкреатит, синдром мальабсорбции с «жирным стулом», «неясные» диспептические нарушения, цирроз печени, сниженная фертильность у женщин, дыхательная недостаточность, «тепловой коллапс». Хронические колиты, холециститы у родственников.
2. Потовый тест: ионофорез с пилокарпином. Повышение Cl более 60 ммоль/л — вероятный диагноз; концентрация Cl > 100 ммоль/л — достоверный диагноз. При этом разница в концентрации хлора и натрия не должна превышать 8-10 ммоль/л. Потовый тест для постановки окончательного диагноза должен быть положительным не менее трех раз. Потовую пробу необходимо проводить каждому ребенку с хроническим кашлем.
3. Химотрипсин в стуле [6]. Химотрипсины — группа эндопептидаз, образуются клетками поджелудочной железы как химотрипсиногены А, В и С. После поступления в желудочно-кишечный тракт активируются. Для определения химотрипсина в стуле необходимо не менее чем за 3 дня до исследования отказаться от приема пищеварительных ферментов. При МВ концентрация химотрипсина в каловых массах снижена. (Проба не стандартизована. Нормативные значения разрабатываются в конкретной лаборатории.) Дифференциальный диагноз проводится с экзокринной панкреатической недостаточностью любой другой этиологии, синдромом Швахмана, состоянием после резекции желудка по Бильрот II, белковым дефицитом.
Ложноотрицательные значения получают при незначительном и умеренном снижении экзокринной функции поджелудочной железы.
Ложноположительные значения могут быть при поздней отмене энзимных препаратов перед проведением пробы.
4. Определение жирных кислот в стуле [6]. В норме менее 20 ммоль/день. Пограничные значения — 20-25 ммоль/день. Проба положительна при снижении экзокринной функции не менее чем на 75%. Дифференциальный диагноз:
- дефицит коньюгированных жирных кислот в тонком кишечнике (как условие эмульгации жиров с их последующей ферментацией липазой) при печеночной недостаточности, непроходимости желчных путей, бактериальной контаминации тонкого кишечника с усиленным гидролизом желчных кислот;
- илеит;
- мальабсорбция при целиакии;
- энтерит;
- интестинальные лимфомы;
- болезнь Уипла;
- пищевая аллергия;
- ускоренный пассаж пищи при диареях, карциноидном синдроме, гипертиреозе.
5. ДНК-диагностика. Наиболее чувствительная и специфическая. Ложные результаты получают в 0,5-3% случаев. Оправданна для стран, где частота дельта-F508 выше 80%. В России из-за относительно низкой частоты последней ДНК-диагностика затруднительна и дорога. Как отмечалось выше, частота разных мутаций варьирует среди этнических групп. Признано, что если ни одна из десяти «локальных» мутаций не обнаруживается на том или ином аллеле хромосомы ребенка и родители его не являются родственниками, вероятность МВ у данного пациента ничтожна. ДНК-обследование родителей и других членов семьи без клинических признаков болезни нецелесообразно, за исключением случаев пренатальной диагностики.
6. Пренатальная ДНК-диагностика. Исследование изоэнзимов интестинальной щелочной фосфатазы из околоплодных вод. Возможно с 18-20 недели беременности. Ложноположительные и ложноотрицательные значения получают в 4% случаев.
7. В настоящее время в нашей стране идентификации доступно около 75% мутантных хромосом, что не позволяет проводить массовый скрининг. Но возможно проведение каскадного скрининга [2], когда в центре находится семья больного МВ и родственники. Скрининг новорожденных может осуществляться методом IRT (иммунореактивный трипсин) или ВМ-лабстик-тест («Хехст»). Метод IRT относительно дорог и дает около 10% ложноположительных и ложноотрицательных результатов. ВМ-тест значительно дешевле, но может дать 15% ложноотрицательных результатов. Скрининг новорожденных представляется перспективным направлением, так как позволяет определить частоту МВ в стране, рано начать интенсивное лечение и идентифицировать семейные пары, нуждающиеся в генетическом консультировании.
- Лечение
Лечение МВ требует комплексного подхода. Еще раз следует подчеркнуть, что оно должно проводиться в специализированных центрах и под их контролем. Лечить ребенка невозможно без тесного сотрудничества с родителями. Поэтому родители должны быть информированы о сути заболевания, характере и особенностях течения процесса, методах лечения, обучены диагностике ухудшений, ряду лечебных и реабилитационных методов. В нашей стране дети с МВ имеют льготы, однако их перечень следует существенно расширить и продлить в возрастном аспекте. Визиты к врачу должны происходить не реже 1 раза в 3 мес. Оценивают антропометрические данные, функцию внешнего дыхания, общие анализы крови и мочи, копрограмму, анализ мокроты на флору и ее чувствительность к антибиотикам. По показаниям проводят рентгенографию грудной клетки, эхографию печени и сердца, исследуют иммунный статус. Желателен «дневной стационар», широко распространенный на Западе и хорошо принятый у нас пациентами и их родителями [2, 13, 15, 16]. Прежде всего вносится коррекция в лечебно-реабилитационный режим. Необходимо эффективно очищать бронхиальное дерево от вязкой мокроты, бороться с инфекцией и обеспечить хорошее физическое развитие больного. Кинезотерапия включает позиционный дренаж, клопф-массаж, вибрацию, специальные дыхательные упражнения, активные циклы дыхания, форсированную экспираторную технику, аутогенный дренаж, дыхание с положительным давлением на выдохе. Последняя процедура проводится под контролем врача, так как возможны осложнения вплоть до пневмоторакса. Обязательно применение бронходилятаторов, муколитиков (см. ниже), по возможности — амилорида (блокатор натрия) и/или «пульмозима» (ДНК-аза, производимая фирмой «Хоффман ля Рош»).
Пациенты с муковисцидозом и члены их семей должны находиться под наблюдением в специализированных центрах. Но в нашей стране таких центров немного, а с учетом расстояний и крайней затруднительности дорожных расходов они становятся и вовсе малодоступными для пациентов. Поэтому принципы ведения больных с МВ должны быть знакомы врачам широкой лечебной сети
Легочная патология. Частое применение антибиотиков. Они должны назначаться при ранних признаках активации воспаления с длительностью курсов до 2-3 недель. Многие антибиотики, эффективные по отношению к псевдомонадам, требуют внутривенного введения. При псевдомонадной инфекции (по антибиотикограмме!) эффективны аминогликозиды, цефалоспорины III поколения, фторхинолоны. Некоторые из цефалоспоринов и тобрамицин могут быть ингалированы. Последний в дозе 300 мг независимо от возраста 1 раз в сутки. При стафилококковом обсеменении эффективен котримаксозол, но клиренс его у пациентов с МВ повышен, поэтому надо увеличить обычную терапевтическую дозу.
Муколитики — непременный атрибут терапии МВ. Назначают как внутрь, так и в ингаляциях: N-ацетилцистеин 300-1200 мг/сутки. (Передозировка препарата ведет к лизису слизистой.) Бронхоскопическое введение муколитиков с последующей аспирацией секрета и антибиотиков в конце процедуры бронхиального лаважа (с дисперсией ультразвуком) — эффективный путь эндоскопического введения препаратов. Современные методики трансназальной гибкой бронхоскопии позволяют применять процедуру амбулаторно.
В случаях бронхоспастического синдрома — ингаляции бета-миметиков (длительное применение чревато развитием аритмий и дилятационной кардиомиопатии), а также кортикостероидов (системно или в ингаляциях) с целью уменьшить воспалительные процессы в легких (побочное действие — остеопороз, избыточный вес, инфекционные осложнения), нестероидных противовоспалительных препаратов. Эти средства снижают воспалительные реакции бронхиального дерева, которые приносят иногда больше вреда, чем собственно инфекционный агент. С этой точки зрения оправданно применение альфа-один-антитрипсина, сывороточного лейкоцитарного ингибитора протеаз, антицитокинов (прежде всего — антиинтерлейкины ИЛ2, ИЛ8). Альтернативные хлоридные каналы открывают АДФ и УДФ (уридиндифосфат).
В странах Северной Америки и в Европе производят пересадку легких или комплекса сердце—легкие, а также разрабатывают генно-инженерные подходы с коррекцией функции мутантного гена путем применения пневмотропных вирусов со встроенными в них генетическими конструкциями. В 1998 г. начата программа генной терапии МВ и в России.
Панкреатическая недостаточность. Хороший нутритивный статус — одна из основных целей в лечении детей с МВ. Поэтому необходима постоянная ферментная терапия. Пациенты с хорошим физическим развитием имеют лучший прогноз. Они более активны, лучше переносят физические нагрузки, имеют лучшие показатели функции внешнего дыхания и иммунитета. Эффективны (по нарастающей) панкреатин, мезим-форте, панзитрат, «Креон». Последние два в виде гранул и микротаблеток с рН-чувствительной оболочкой. Доза индивидуальна. Обычно начинают с 2–6 тыс ед. липазы на кг массы/сутки. Повышают постепенно, исходя из характеристик стула, показателей веса ребенка. Превышение дозы приводит к раздражению слизистой кишечника, воспалению. В крайне редких случаях возможны поствоспалительные стриктуры толстой кишки. Хороший эффект при поражении печени (холестаз, предцирроз, цирроз) оказывает назначение урсодезоксихолевой кислоты (урсосан) в сочетании с таурином, способствующим выведению желчных кислот, которые облегчают переваривание жиров.
Принципы ведения пациента, контрольные исследования. Питание должно превышать возрастные калорийные нормы на 10–15%, обязательно введение поливитаминов, микроэлементов. Белковая диета без ограничения жиров, но при адекватной заместительной терапии современными микросферическими ферментами с рН-чувствительной оболочкой.
Контроль массы тела. Снижение веса или плоская весовая кривая указывают на плохое ферментное обеспечение или на обострение хронического бронхолегочного процесса.
Бактериологическое исследование мокроты с антибиотикограммой или мазка из зева 1 раз в 6 месяцев и после обострения бронхолегочного процесса или при изменении цвета мокроты (зеленый цвет, примесь крови).
HbA1 — гликолизированный гемоглобин. У детей старше 8 лет определять 1-2 раза в год. При сниженной толерантности к глюкозе — чаще.
Рентгенографию органов грудной клетки делать при обострении бронхолегочного процесса, особенно при подозрении на пневмонию. Как контроль — 1 раз в год.
Эхокардиограмма (особенно правых отделов, легочной артерии) не реже 1 раза в год.
ЭКГ. 1–2 раза в год. По показаниям — чаще.
Функциональные пробы легких. Функция внешнего дыхания (обычно — с 6 лет, возраста кооперативного участия) и газы крови — 1 раз в месяц и после обострения бронхолегочного процесса.
Бодиплетизмография. С 8 лет — 1-2 раза в год. По показаниям — чаще. Функцию внешнего дыхания не исследуют в терминальной стадии, так как в префинальном периоде тесты обременительны для больного и не сказываются на лечении.
При подозрении на цирроз печени — ультразвуковые исследования, печеночные функции, протромбин, реже — биопсия.
- Осложения и исходы
Полипы носа. Стероиды ингаляционно или в виде мазевых аппликаций. Оперативное лечение нецелесообразно (вероятность рецидивов высока).
Пневмоторакс. Развивается у детей старшего возраста и взрослых. Вероятность рецидивирования высока. Необходимы покой, при объеме менее 10% легочного — минимум манипуляций. При напряженном пневмотораксе — дренаж, плевральная пункция. При рецидивах — удаление буллезной доли, плевродез.
Ателектазы. Необходимы бронхоскопии с бронхоальвеолярным лаважем и введением муколитиков, антибиотиков, дыхательная гимнастика.
Пневмонии. Общие принципы терапии. Крайне важны дренирующие мероприятия.
Кровохарканье. Как симптом переоценивается и вызывает неоправданную панику. Выглядит как примесь крови к мокроте, чаще всего обусловлено поражением слизистой бронхов. При легочном кровотечении (300 и более мл одномоментно или более 100 мл за 3 дня) — ангиографическая эмболизация или окклюзия кровоточащего сосуда. При неудаче — перевязка сосуда или резекция сегмента (доли).
Легочное сердце. При адекватной терапии развивается как манифестная форма только у взрослых. Им же свойственны и нарушения ритма сердца. Дигиталис при «легочном сердце» малоэффективен, желательно применение коринфара, нифедипина и гидралазина (последний опасен возможными аутоиммунными реакциями).
Аспергиллез. Ассоциируется с кортикостероидной терапией. Если аспергиллы обнаружены в мокроте случайно и не проявляются клинически, то лечения не требуется. Терапия назначается при распространенных бронхоэктазах, расширении бронхов, нарастании легочной симптоматики, особенно с признаками торпидной обструкции, повышении общего IgЕ и специфического IgE.
Аллергия и астма. У 25–48% больных наблюдается сочетание МВ и астмы.
Соледефицитный эксикоз. Может быть не только у новорожденных, но и у детей разного возраста и у взрослых, особенно в жаркое время года. Профилактика — обильное питье и достаточное поступление солей (3–8 г/сутки).
Сахарный диабет панкреатогенный. Развивается очень медленно, постепенно. Наблюдается у 2% детей и у 15% взрослых, больных МВ.
Кровотечение желудочное и из варикозных вен пищевода (при циррозе печени). Проводятся эндоскопическое склерозирование варикозно расширенных вен, частичная резекция селезенки, шунтирующие операции.
Камни желчного пузыря. Эндоскопическое удаление менее рискованно, чем лапаротомическое, при котором выше вероятность легочных осложнений.
Эквивалент мекониального илеуса у подростков и взрослых — частичная дистальная интестинальная обструкция с густым вязким стулом. При илеусе, не требующем хирургического вмешательства, — промывание гастрографином, гипаком, N-ацетилцистеином. При отсутствии эффекта — хирургическое вмешательство.
Пневматоз стенки кишки может быть обнаружен случайно и сам по себе не требует вмешательства.
Выпадение прямой кишки при адекватной ферментной заместительной терапии встречается очень редко.
Легочная остеоартропатия. Наряду с деформациями концевых фаланг могут появиться периоститические боли в длинных трубчатых костях. Для облегчения состояния назначают нестероидные противовоспалительные средства.
Деформации грудной клетки развиваются как итог легочной патологии.
Таким образом, объединенные усилия педиатров, терапевтов, бронхологов, гастроэнтерологов, специалистов по лечебному питанию, психологов, социальных работников при условии ранней диагностики и адекватной терапии, заинтересованном участии в лечении пациента и его родственников уже на сегодня способствуют изменению качества жизни и увеличивают ее продолжительность у больных МВ.
Литература
1. Капранов Н. И., Рачинский С. В. Муковисцидоз. М., 1995, с. 188.
2. Капранов Н. И., Каширская Н. Ю. Актуальные проблемы муковисцидоза. Педиатрия, 1998, № 1, с. 61-66.
3. Консенсунс по муковисцидозу. Медицинская газета, 1995, № 41, 02.06.95.
4. Петрова Н. В. Материалы научно-практической конференции РДКБ, М., 1995, с. 96.
5. Потапова О. Ю. Молекулярно-генетический анализ кистозного фиброза в России. Автореферат к. м. н., СПб, 1994, с. 24.
6. Becker D. Vademecum Labordiagnostic. Berlin, 1987, ss. 334.
7. Dean T., Dai Y., Shute K., e. a. Pediatric Research, 1993, Vol. 34, p. 159-161.
8. Dodge J., Brock D., Widdicombe J. Cystic Fibrosis. UK, 1994, p. 3.
9. Fanconi G., Uelinger E., Knauer C. Das Coeliakie-Syndrom bei angeborener zystischer Pancreasfibrose und Bronchiektasien. Wiener med. Wochenshrift, 1936, Bd. 86, ss. 753-756.
10. Fick R., Rabbins R. Pediatric Research, 1995, Vol. 20, p. 1258.
11. Hodson M., Geddes D. Cystic fibrosis. London, 1995, p. 439.
12. Illing St. Mukoviszidose. В кн. Paediatrie. Stuttgart, 1993, ss. 437-440.
13. Kopelman H., Davies M. Cystic fibrosis. in: Current Pediatric Therapy, Philadelphia, 1993, 4-th Ed. pp. 135-139.
14. Leiber G. Die klinischen Syndrome. Muenchen, 1993, Bd. 1,2.
15. Welsh M., Smith A. Cystic fibrosis, N.Y., 1995, p. 36-43.
16. WHO/HGN/IGF(M)A. Guidelines for the Diagnosis and Mangement of CF., Geneve, 1996, p. 59.