- Research
- Open Access
- Published: 09 April 2021
Italian Journal of Pediatrics
volume 47, Article number: 87 (2021)
Cite this article
-
2016 Accesses
-
1 Citations
-
1 Altmetric
-
Metrics details
Abstract
Background
Cystic Fibrosis newborn screening (CFNBS) is the optimal method to diagnose the disease during the asymptomatic period. The aim of the study was to determine how CFNBS affects long term clinical outcomes.
Methods
Data from infants who were born in Lodz Voivodship, referred to CF center as a part of CFNBS according to IRT/DNA protocol were compared to the data of children with established CF diagnosis before the start of NBS in Poland (Group CF, n = 52).
Results
In 37 children (during 151 referred infants) the diagnosis of CF was established due to CF NBS (CF NBS Group, n = 37). The average time of diagnosis was 1.59 month in Group CF NBS and 45.25 months in 52 children from Group CF.
Pulmonary exacerbations occurred on average 4.2 times in Group CFNBS and they were hospitalized on average 0.5 times compared to Group CF – respectively 6.77 and 2.14 (p < 0.001).
The number of PA infected patients increased between the fifth and eighth year of age (OR = 1.16 (95% CI: 1.04–19) (P = 0.007)) regardless of the study group (P = 0.984). Patients with MRSA infection have a higher risk of PA infections in subsequent years of their life (OR = 1.45 (95% CI: 1.03–2.03) (P = 0.032)).
Conclusions
CF NBS has beneficial effects primarily on decrease of pulmonary withhope for a longer life expectancy and better and centralised treatment in multidisciplinary CF focused centres.
Introduction
Cystic fibrosis (CF) is the most common fatal autosomal recessive disease in Caucasians [1]. It is widely recognized that CF is a variable condition, that may affect the respiratory tract, pancreas, intestine, male genital tract, hepatobiliary system and exocrine sweat glands, resulting in complex multisystem disease. The severity of clinical symptoms results due to pathogenic mutations in both alleles of the CF transmembrane conductance regulator (CFTR) gene. More than 1950 mutations have been described so far in the CFTR gene, grouped in 5 classes based on the impact on protein synthesis or activity [2, 3].
Over the last 50 years, European countries have introduced newborn bloodspot screening (NBS) for a range of inherited diseases as an important public health program. In 2004, the European CF Society established the Neonatal Screening Working Group (NSWG) to support the implementation of CF NBS program across Europe [4, 5]. CF NBS is the optimal method to diagnose the disease during the asymptomatic period. The primary goal of newborn screening is to decrease morbidity, mortality and associated disabilities. The early diagnosis is associated with improved physical development and slower lung deterioration due to the lower frequency of infections.
All CF NBS programs begin with detection of an elevated immunoreactive trypsinogen (IRT) level in a dried blood specimen from the newborn [3, 5, 6]. A positive IRTscreen is triaged to second-tier testing, which is DNA mutation testing in many national screening programs, including Poland (IRT/DNA protocol) [3,4,5,6,7]. Despite the advent of NBS and improved knowledge about CFTR genetics, CF diagnosis remains incomplete for many reasons, such as inconclusive sweat chloride values, CFTR mutations of uncertain pathogenicity, and differential expression of CFTR or modifier effects [8]. Also CF NBS introduced a new complexity and diagnostic dilemma, namely infants with abnormal screening tests, because of elevated immunoreactive IRT levels but inconclusive sweat tests and/or DNA results. The term for infants with an inconclusive diagnosis have been proposed: CF screen positive, inconclusive diagnosis (CFSPID) in Europe [9].
The first pilot NBS CF study was performed in Poland from 1999 to 2002 and included about 25% of the Polish population. The current program was implemented in 2006 and was expanded to the whole country in the summer of 2009. In Lodz Voivodeship, CF NBS was introduced on 1st June 2009 [3, 6, 7].
The research presents 10 years of experience of our CF center with the national newborn CF screening program in Lodz Voivodship and including the analyzis of diagnostic dilemma occurred during the program. The aim of the study was to determine how early diagnosis of CF, using neonatal screening with IRT/DNA protocol, affects long term clinical outcomes in comparison to CF patients with disease diagnosed before CF NBS?
Material and methods
Patients
Patients participated in the screening program
The study population comprised of infants who were born between 01.07.2009 and 31.12.2019 in Lodz Voivodship and were referred to our CF center as a part of NBS for CF and performed CFTR gene analysis according to IRT/DNA protocol described below. The infants had been evaluated at least once per 3 months. They were divided to three groups according to laboratory tests and further clinical evaluation as CF NBS Group, CF SPID and false positive NBS. Cystic fibrosis (CF NBS Group) was diagnosed in infants based on the level of sweat chloride in addition to evidence of CFTR dysfunction. The designation CF SPID was established to asymptomatic infants if they presented a positive CF NBS test plus: sweat chloride < 59 mmol/L and 2 CFTR mutations with 0–1 CF-causing CFTR mutations. The asymptomatic CF NBS positive infant with presence of no CFTR mutation or mutation in one allele plus a negative sweat test, referred as false positive NBS (including carriers).
Patients diagnosed with CF before introduction of screening program
We also analyzed data of 52 patients (24 females and 28 males) with established CF diagnosis born between 1996 and 2009 (before the newborn screening in Poland) attending our Cystic Fibrosis Outpatient Center in Copernicus Hospital in Lodz, Poland. Patients in this group had been referred to the Center due to occurrence of symptoms suggesting CF (CF Group).
Next, we compared both groups CF NBS and CF. We have chosen the interval — between 5 and 8 years of age to compare both groups, because at that time the most data were available.
Data assessment
In both groups the data were obtained retrospectively after a review of patient charts. The socio-economic data were analysed. These included a place of living defined as a large city above 100,000 inhabitants, a small city 10,000–100,000 inhabitants, and a village below 10,000 inhabitants. The perinatal period contained data: delivery data and weight, Apgar score and presence of meconium ileus (MI), time of CF diagnosis. We also analysed body weight and percentile of body weight at 5 and 8 years of age.
Clinical evaluation
Respiratory status was assessed by the forced expiratory volume in 1 s (FEV1) in spirometry, expressed as percentages of predicted values for height, weight, age, and gender. All analyzed measurements were done in fifth and 8 year of life, only in stable periods (using a Jaeger MasterScreen Body spirometer; E Jaeger GmbH; Wurzburg, Germany). The tests were performed according to American Thoracic Society standards [10].
The number of bronchopulmonary exacerbations and number of hospitalizations (due to pulmonary disease, longer than 3 days) between five and 8 years of life were evaluated. Pulmonary exacerbation was defined as excessive sputum expectoration, general malaise, and need for antibiotics. Chronic bacteria colonization Pseudomonas aeruginosa (PA) or methicillin-resistant Staphylococcus aureus (MRSA) were also collected, and defined as three positive consecutive sputum cultures over a period of 6 months in fifth and 8 year of life.
CF NBS protocol
IRT was measured in dried blood samples from 3 to 5 days old neonates. The IRT concentration cut-off was established as > 99.4 percentile (according to the pilot CF NBS program [6]). The elevated IRT caused further genetic testing from the same dried blood samples. In neonates with meconium ileus (MI), the DNA analysis was performed regardless of IRT level. The measurements of IRT (by using IRT Neonatal Screening ELISA colorimetric assay — IBL International) and DNA analysis from sampling paper (processing with the Extract Blood PCR Kit) were performed in the Genetic Department, Institute of Mother and Child, Warsaw, Poland. Since September 2011, the extended DNA analysis panel has been used and comprised 95% of mutated alleles in the Polish population.
Neonates with one or two mutations in CFTR detected due to NBS examinations were directed to CF Centres for clinical assessment and sweat tests (measured by the quantitative pilocarpine iontophoresis method and Nanoduct method parallel) [6]. CF NBS was conducted as a pilot programme in four Polish districts in the period 1999–2003. In 2006 CF NBS started again in the same regions and was gradually extended across the country. In June 2009 covered all of newborn population in Poland. In the 1980s, the prevalence of CF for Caucasians was estimated at 1: 2500 live births. The disease incidence was more accurately calculated to 1: 4000–5000 thanks to the extensive implementation of CF NBS in the world. In Poland due to data from CF NBS in the years 2006–2010 it is believed that one child every 4394 live births is born with CF.
The study was approved by the Ethical Committee of the Medical University of Lodz, Poland (nr RNN/145/20/KE).
Statistical analysis
Numerical traits were described by way of the arithmetic mean, standard deviation (SD), standard error (SE), when applicable, and their minimum-to-maximum values. Categorical variables were depicted by using counts (n) and percentages (%).
Multiple logistic (for binary dependent variables), multiple linear (for numerical traits), and multiple Poisson (for count data) regression models were fitted in order to test statistical co-dependencies. When dealing with non-normally distributed variables, robust standard errors (i.e. sandwich estimators) were used in the regression equations. All the regression models were controlled for the studied patients’ characteristics such as sex, gender, place of residence, birth weight, APGAR and also for crucial infections (PA, MRSA) before their 5th year of age. Missing data were case-wise deleted.
A level of P < 0.05 was deemed statistically significant. All the computations were performed by using of Stata/Special Edition, release 14.2 (StataCorp LP, College Station, Texas, USA).
Results
Evaluation of patients participated in the screening program (CF NBS positive infants)
One hundred fifty one infants were referred to our CF center as a part of NBS for CF due to CF suspicion: 84 females and 67 males. The CFTR gene analysis for each positive IRT, and neonates with MI regardless of IRT levels, allowed to identify mutations in one allele in 145 patients and in both alleles in 44 patients (96.03% of screened CF patients). Among 44 cases, after biochemical, genetic and clinical evaluations, in 37 children the diagnosis of CF was established (24.5%) during the first verifying visit (CF NBS Group) — in 76% the diagnosis was establish up to 35 days after birth, in 9 patients the first verifying visit took place later in life due to hospitalization connected with their MI. The mean birth weight in CF NBS patients was 3226.34 g, In this group, 9 patients had MI (24.32%). In the remaining 7 (out of 44) cases (15.9%), despite detection of two CFTR mutations, a clinical evaluation did not confirm the CF diagnosis (CF SPID). These infants had the CFTR genotype as follows: 3 patients with [F508del]; [IVS8-5 T+(TG)11], [3849 + 10kbC > T];[R117H], [Y301C];[3271 + 18C > T], [V754M];[V562L], [F508del];[R117H]. The above mutations found at least in one allele causing unproven or uncertain clinical consequences. These patients remain under the observation of our clinic. The most common mutation was F508 del, detected in 104 patients in one or both alleles (68.88%).
Six patients (3.97%) who were directed to the Cystic Fibrosis Outpatient based on the IRT elevations, had DNA analysis for CF mutations negative and they sweat chloride test was correct (false positive NBS). After further observations and examinations, the CF was excluded. So far (until December 2019) we also have information about two cases of false-negative results in CF NBS in our region (false negative NBS).
Comparison of group CF NBS and group CF
At the time of data collection 27 of CF NBS patients were at least 8 years old, remain under the medical care in our CF Outpatient Center and were included into compared analysis. The Group CF NBS with positive result of newborn CF screening included: 59% females, mean birth weight 3168.52 g and mean Apgar 8.78. Mutation F508 del was detected in both alleles in 29.63% patients.
Fifty two patients belonging to the Group CF, were born between 1996 and 2009 (before the start of newborn screening in Poland) – 24 females and 28 males. They were included into compared analysis with Group CF NBS. Their mean birth weight was 3195.6 g, Apgar 8.54, F508 del mutation in both alleles was detected in 50% cases.
Group CF NBS and Group CF demographic and clinical characteristics are shown in Table 1.
Full size table
The average time of the CF diagnosis was 45.25 months from birth in Group CF.
and 1.59 months in Group CF NBS.
The most significant difference between this two Groups was the number of exacerbations and hospitalizations between 5 and 8 years of life (p < 0.001). Pulmonary exacerbation in analysed period occurred on average 4.2 times in children in Group CF NBS and they were hospitalized on average 0.5 times. While in Group CF children mean of pulmonary exacerbations was 6.77 and mean number of hospitalization was 2.14.
Even though the average body weight and its percentile value was higher in the Group CF NBS, this difference was not significant. What is more weight gain between 5 and 8 years old was greater in the Group CF. No statistically significant predictors affecting weight gain in both groups were defined.
In spirometry measurements patients in both groups presented stable values of FEV1 during at least 3 years of observation, and the difference was not significant.
The number of PA infected patients increased significantly between the fifth and eighth year of age of the examined children (OR = 1.16 (95% CI: 1.04–19) (P = 0.007)) regardless of the study group (P = 0.984). It seems that patients with MRSA infection in the fifth year of life have a higher risk of PA infections in subsequent years of their life (OR = 1.45 (95% CI: 1.03–2.03) (P = 0.032)). The number of patients with PA or MRSA infection in the fifth year of life and PA infection in the age of 8 was greater in the CF Group, but these differences were not statistically significant.
Descriptive characteristics of all CF patients by screening status are shown in Table 2.
Full size table
Discussion
According to the Neonatal Screening Working Group new-born screening for CF provides an immediate diagnosis, before the onset of clinical symptoms [11,12,13]. In the beginning CF NBS raised doubts about ethical aspects with regard to possible benefits and risks. After many years of experience CF NBS has been widely implemented and accepted. Ten years of CF NBS programme in Lodz Voivodship leads to diagnosis of CF in 37 neonates among 151 children directed after their birth with CF suspicion to our Outpatient Center. The incidence of CF in Poland based on neonatal screening is currently 1:4394–1:5000 [3, 6].
CF NBS has been based on the assumption that pre-symptomatic detection permits early access to specialised medical care, and thus results in less morbidity and longer life expectancy. In our region CF NBS screened patients were diagnosed and treated in multidisciplinary CF centre between first and second month of their life. For comparison the median age of the diagnosis of patients, who were born before the start of CF NBS in Poland due to typical CF symptoms – 45.25 months from their birth. According to the literature, the most benefits associated with early identification of CF including include better growth and lung function, less intensive therapeutic burden and reduced cost of care [11,12,13,14]. Our observations show that early diagnosis and the introduction of appropriate treatment for asymptomatic CF patients resulted in a lower frequency of the pulmonary exacerbation and lower number of hospitalizations. It is very beneficial observation due to reports which emphasise that the repeated mild-to-moderate pulmonary exacerbations, especially in the first years of life result in airway remodelling. While more severe hospitalised episodes could increase structural airway injury risk [13,14,15,16]. Data from Australia showed improved survival in future in patients with lower number of hospitalisations in the first 3 years of life [17]. The occurrence of respiratory exacerbations are associated with an accelerated decline in lung function and reduced quality of life and survival [15,16,17]. On the other hand mean values of FEV1 in the analysed patients, presented stable results during at least 3 years of observation; there was no difference between Groups. This may be due to the age — below 8 — too early to identify significant differences in FEV1 secondary to clinical outcome (such as pulmonary exacerbations, bacterial colonisations), which likely takes longer to cause impairment of lung function that is identifiable with spirometry [16].
Children with CF are highly susceptible to chronic respiratory tract colonization and subsequent recurrent infection. It is know that once chronic PA infection is established, the risk of mortality and morbidity increases [18, 19]. In our study the mean number of patients with PA or MRSA colonization in the 5 years old and PA chronic infection in the age of 8 was lower among children screened in NBS, but there were no significant differences between both groups. The number of PA infected persons increased significantly between the fifth and eighth year of age regardless of belonging to the study group. Although the increasing number of SA and PA colonized patients were observed with age in both CF NBS and CF group, the CF NBS children had fewer exacerbations and hospitalisations due to intensively treatment from the very first PA sputum detection. The important observation was that patients in both groups with SA infection in the fifth year of life have a higher risk of PA infections in subsequent years of their life. Persistent presence of MRSA in the respiratory tract is associated with increased rates of PA through mediation of PA biofilm formation [18, 19]. In practice, we suggest to employ intermittent, symptom-based treatment of SA especially MRSA to delay initial PA colonization. The colonization with PA remains the most ordinary airway pathogen contributing to shortened survival in CF patients. For over a two decades antibiotic effective for PA (for example tobramycin inhalation solution (TIS)) is recommended for the treatment and early introduction with TIS has resulted in improved lung function and reduced risk of airway exacerbations [20, 21]. In our study in CF group the diagnosis was established mean in 42.25 month of life, but even maximally in 180 months of life. In such situation at the time of CF recognition patients’ sputum were often positive for PA and we treated PA colonization not an initial infection. Patients from CF NBS group were treated intensively from the very first PA sputum detection. We believe this may have contributed to a reduction in the number of pulmonary exacerbations and associated hospitalizations. Moss et al. [21] proved that delay in initiation of TIS therapy is associated with reduced long-term improvement over baseline, suggesting an irreversible component to lung function decline. To be maximally effective, TIS treatment should begin early after first detection of PA. In agreement to this study, although the incidence of PA found in the airways became similar over the years in both study groups (CF and CF NBS) the number of exacerbations and hospitalizations in CF NBS group was significant lower.
CF NBS allows the affected infants to receive immediate treatments including not only physiotherapy and preventing of pulmonary manifestations of the disease, but also great emphasis is pleased on assessment of nutritional status, and pancreatic enzyme replacement therapy. The Wisconsin randomised trial on newborn screening for CF showed that screened neonates exhibit better nutritional status in the first years of life [22], but no output is available from this cohort on long-term survival. We observed trend that body weight were higher in the CF NBS Group and they presented systematic and stable weight gain but this results were not statistically significant.
According to data published by Bobadilla et al., the complete CFTR gene analysis by sequencing of selected regions should identify mutations in one or both alleles in nearly 95% of screened children [23] and IRT/DNA strategy to achieve sensitivity close to 100% (despite children with MI, whose IRT level is not elevated). Due to Fritz formula the predicted false-negative rate for the Polish population is 6–7 cases per year, which gives 1–2 omitted CF cases per 100,000 live births [24]. It shows that despite the high sensitivity and specificity of IRT/DNA protocol it is important to follow patients with CF suggestive symptoms even when CF NBS was negative. We had two cases of false-negative results in CF NBS in our region to this day. According to normal values of the IRT in new-borns with MI they are in risk group of a false-negative first step of CF NBS protocol. Each case of MI should be reported to the NBS center by appropriate note on sampling paper and neonates must to be verify in CF centers. In our study, the new-borns with MI represent 24% of CF diagnosis, while the literature data shows MI frequency about 10–20% [25].
The IRT/DNA strategy allows to detect not only CF disease but also carries of the CFTR gene mutations, as well as cases of unclear clinical consequences. This fact constitutes a definite psychological problem for patients and their parents and raised doubts about ethical aspects of the CF NBS. It may have negative influences on further procreation of such persons. We detected 101 patients with mutations only in one allele of CFTR gene (what was 66.89% of all neonates from CF NBS). They were defined as carriers and in all cases, a genetic consultations for their parents and siblings was recommended.
Some limitations of our current study include a single centre analysis, which limits its generalizability. What is more we made the comparison to historical data (a blinded controlled study with CF- newborns left untreated was unacceptable due to ethical reasons).
Over the years new therapies were approved, standards of care established therefore, the outcomes of same patients in subsequent years is expected to be improved, even without the advent of newborn screening. We have to choose the appropriate time interval to compere a historical cohort (CF Group) and CF NBS positive patients.
Conclusion
In conclusion, newborn screening contributes to early diagnosis of cystic fibrosis. The presented study showed that CF NBS has beneficial effects primarily on decrease of pulmonary exacerbations with hope for a longer life expectancy in these group.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CF:
-
Cystic fibrosis
- CFNBS:
-
Cystic Fibrosis newborn screening
- CFTR:
-
CF transmembrane conductance regulator
- FEV1:
-
The forced expiratory volume in 1 s
- IRT:
-
Immunoreactive trypsinogen
- MI:
-
Meconium ileus
- NSWG:
-
Neonatal Screening Working Group
- PA:
-
Pseudomonas aeruginosa
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- TIS:
-
Tobramycin inhalation solution
References
-
Farrell PM. The prevalence of cystic fibrosis in the European Union. J Cyst Fibros. 2008;7(5):450–3. https://doi.org/10.1016/j.jcf.2008.03.007.
Article
PubMedGoogle Scholar
-
Paranjapye A, Ruffin M, Harris A, Corvol H. Genetic variation in CFTR and modifier loci may modulate cystic fibrosis disease severity. J Cyst Fibros. 2019;13(19):30963–4.
Google Scholar
-
Sobczyńska-Tomaszewska A, Ołtarzewski M, Czerska K, Wertheim-Tysarowska K, Sands D, Walkowiak J, et al. Newborn screening for cystic fibrosis: polish 4 years experience with CFTR sequencing strategy. European J Human Genet. 2013;21(4):391–6. https://doi.org/10.1038/ejhg.2012.180.
Article
CASGoogle Scholar
-
Barben J, Castellani C, Dankert-Roelse J, Gartner S, Kashirskaya N, Linnane B, et al. The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe. J Cyst Fibros. 2017;16(2):207–13. https://doi.org/10.1016/j.jcf.2016.12.012.
Article
PubMedGoogle Scholar
-
Loeber JG, Burgard P, Cornel MC, Rigter T, Weinreich SS, Rupp K, et al. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 1. From blood spot to screening result. J Inherit Metab Dis. 2012;35(4):603–11. https://doi.org/10.1007/s10545-012-9483-0.
Article
PubMedGoogle Scholar
-
Sands D, Zybert K, Mierzejewska E, Ołtarzewski M. Diagnosing cystic fibrosis in newborn screening in Poland — 15 years of experience. Dev Period Med. 2015;19(1):16–24.
PubMed
Google Scholar
-
Zietkiewicz E, Rutkiewicz E, Pogorzelski A, Klimek B, Voelkel K, Witt M. CFTR mutations spectrum and the efficiency of molecular diagnostics in polish cystic fibrosis patients. PLoS One. 2014;26(2):8909.
Google Scholar
-
Levy H, Nugent M, Schneck K, Stachiw-Hietpas D, Laxova A, Lakser O, et al. Refining the continuum of CFTR-associated disorders in the era of newborn screening. Clin Genet. 2016;89(5):539–49. https://doi.org/10.1111/cge.12711.
Article
CAS
PubMed
PubMed CentralGoogle Scholar
-
Ren C, Borowitz D, Gonska T, Howenstine M, Levy H, Massie J, et al. Cystic fibrosis transmembrane conductance regulator-related metabolic syndrome and cystic fibrosis screen positive, inconclusive diagnosis. J Pediatr. 2017;181:45–51.
Article
Google Scholar
-
Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi A, Coates A, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–38. https://doi.org/10.1183/09031936.05.00034805.
Article
CASGoogle Scholar
-
Castellani C, Massie J, Sontag M, Southern KW. Newborn screening for cystic fibrosis. Lancet Respir Med. 2016;4(8):653–61. https://doi.org/10.1016/S2213-2600(16)00053-9.
Article
PubMedGoogle Scholar
-
Tridello G, Castellani C, Meneghelli I, Tamanini A, Assael BM. Early diagnosis from newborn screening maximises survival in severe cystic fibrosis. ERJ Open Res. 2018;4:2.
Article
Google Scholar
-
Castellani C, Linnane B, Pranke I, Cresta F, Sermet-Gaudelus I, Peckham D. Cystic fibrosis diagnosis in newborns, children, and adults. Semin Respir Crit Care Med. 2019;40(6):701–14. https://doi.org/10.1055/s-0039-1697961.
Article
PubMedGoogle Scholar
-
Barben J, Castellani C, Dankert-Roelse J, Gartner S, Kashirskaya N, Sands D. Et all. The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe. J Cyst Fibros. 2017;16(2):207–13. https://doi.org/10.1016/j.jcf.2016.12.012.
Article
PubMedGoogle Scholar
-
Byrnes C, Vidmar S, Cheney J, Carlin J, Armstrong D, Cooper P, et al. Prospective evaluation of respiratory exacerbations in children with cystic fibrosis from newborn screening to 5 years of age. Thorax. 2013;68(7):643–51. https://doi.org/10.1136/thoraxjnl-2012-202342.
Article
PubMed
PubMed CentralGoogle Scholar
-
Goss C. Acute pulmonary exacerbations in cystic fibrosis. Semin Respir Crit Care Med. 2019;40(6):792–803. https://doi.org/10.1055/s-0039-1697975.
Article
PubMed
PubMed CentralGoogle Scholar
-
Reid DW, Blizzard CL, Shugg DM, Flowers C, Cash C, Greville HM. Changes in cystic fibrosis mortality in Australia, 1979-2005. Med J Aust. 2011;195(7):392–5. https://doi.org/10.5694/mja10.11229.
Article
PubMedGoogle Scholar
-
Mostofian F, Alkadri J, Tang K, Thampi N, Radhakrishnan D. A real world evaluation of the long-term efficacy of strategies to prevent chronic Pseudomonas aeruginosa pulmonary infection in children with cystic fibrosis. Int J Infect Dis. 2019;85:92–7. https://doi.org/10.1016/j.ijid.2019.05.026.
Article
PubMedGoogle Scholar
-
Armbruster CR, Wolter DJ, Mishra M, Hayden HS, Radey MC, Merrihew G, et al. Staphylococcus aureus protein A mediates interspecies interactions at the cell surface of Pseudomonas aeruginosa. MBio. 2016;7:3.
Article
Google Scholar
-
Sawicki GS, Signorovitch JE, Zhang J, Latremouille-Viau D, vonWartburg M, Wu EQ, et al. Reduced mortality in cystic fibrosis patients treated with tobramycin inhalation solution. Pediatr Pulmonol. 2012;47(1):44–52. https://doi.org/10.1002/ppul.21521.
Article
PubMedGoogle Scholar
-
Moss RB. Long-term benefits of inhaled tobramycin in adolescent patients with cystic fibrosis. Chest. 2002;121(1):55–63. https://doi.org/10.1378/chest.121.1.55.
Article
CAS
PubMedGoogle Scholar
-
Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin cystic fibrosis neonatal screening study group. Pediatrics. 2001;107(1):1–13. https://doi.org/10.1542/peds.107.1.1.
Article
CAS
PubMedGoogle Scholar
-
Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations – correlation with incidence data and application to screening. Hum Mutat. 2002;19(6):575–606. https://doi.org/10.1002/humu.10041.
Article
CAS
PubMedGoogle Scholar
-
Fritz A, Farrell P. Estimating the annual number of false negative cystic fibrosis newborn screening tests. Pediatr Pulmonol. 2011;47(2):207–8. https://doi.org/10.1002/ppul.21561.
Article
PubMedGoogle Scholar
-
Tan SMJ, Coffey MJ, Ooi CY. Differences in clinical outcomes of paediatric cystic fibrosis patients with and without meconium ileus. J Cyst Fibros. 2019;18(6):857–62. https://doi.org/10.1016/j.jcf.2019.09.008.
Article
PubMedGoogle Scholar
Download references
Acknowledgements
Not applicable.
Author information
Authors and Affiliations
-
Department of Pediatrics and Allergy, Medical University of Lodz, Copernicus Memorial Hospital, Korczak Paediatric Center, Piłsudskiego 71 Str, 90-329, Lodz, Poland
M. Olszowiec-Chlebna, E. Mospinek & J. Jerzynska
Authors
- M. Olszowiec-Chlebna
You can also search for this author in
PubMed Google Scholar - E. Mospinek
You can also search for this author in
PubMed Google Scholar - J. Jerzynska
You can also search for this author in
PubMed Google Scholar
Contributions
M. Olszowiec-Chlebna MD PhD (literature search, study design, analysis of data, manuscript preparation), E. Mospinek MD (literature search, analysis of data). J. Jerzynska MD, PhD, Prof (study design, manuscript preparation, review of manuscript). The author(s) read and approved the final manuscript.
Corresponding author
Correspondence to
J. Jerzynska.
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethical Committee of the Medical University of Lodz, Poland (nr RNN/145/20/KE).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and Permissions
About this article
Cite this article
Olszowiec-Chlebna, M., Mospinek, E. & Jerzynska, J. Impact of newborn screening for cystic fibrosis on clinical outcomes of pediatric patients: 10 years’ experience in Lodz Voivodship.
Ital J Pediatr 47, 87 (2021). https://doi.org/10.1186/s13052-021-01040-5
Download citation
-
Received: 03 November 2020
-
Accepted: 26 March 2021
-
Published: 09 April 2021
-
DOI: https://doi.org/10.1186/s13052-021-01040-5
Keywords
- Cystic fibrosis
- New born screening
- Diagnosis
- IRT/DNA protocol
- Children
- Research
- Open Access
- Published: 09 April 2021
Italian Journal of Pediatrics
volume 47, Article number: 87 (2021)
Cite this article
-
2016 Accesses
-
1 Citations
-
1 Altmetric
-
Metrics details
Abstract
Background
Cystic Fibrosis newborn screening (CFNBS) is the optimal method to diagnose the disease during the asymptomatic period. The aim of the study was to determine how CFNBS affects long term clinical outcomes.
Methods
Data from infants who were born in Lodz Voivodship, referred to CF center as a part of CFNBS according to IRT/DNA protocol were compared to the data of children with established CF diagnosis before the start of NBS in Poland (Group CF, n = 52).
Results
In 37 children (during 151 referred infants) the diagnosis of CF was established due to CF NBS (CF NBS Group, n = 37). The average time of diagnosis was 1.59 month in Group CF NBS and 45.25 months in 52 children from Group CF.
Pulmonary exacerbations occurred on average 4.2 times in Group CFNBS and they were hospitalized on average 0.5 times compared to Group CF – respectively 6.77 and 2.14 (p < 0.001).
The number of PA infected patients increased between the fifth and eighth year of age (OR = 1.16 (95% CI: 1.04–19) (P = 0.007)) regardless of the study group (P = 0.984). Patients with MRSA infection have a higher risk of PA infections in subsequent years of their life (OR = 1.45 (95% CI: 1.03–2.03) (P = 0.032)).
Conclusions
CF NBS has beneficial effects primarily on decrease of pulmonary withhope for a longer life expectancy and better and centralised treatment in multidisciplinary CF focused centres.
Introduction
Cystic fibrosis (CF) is the most common fatal autosomal recessive disease in Caucasians [1]. It is widely recognized that CF is a variable condition, that may affect the respiratory tract, pancreas, intestine, male genital tract, hepatobiliary system and exocrine sweat glands, resulting in complex multisystem disease. The severity of clinical symptoms results due to pathogenic mutations in both alleles of the CF transmembrane conductance regulator (CFTR) gene. More than 1950 mutations have been described so far in the CFTR gene, grouped in 5 classes based on the impact on protein synthesis or activity [2, 3].
Over the last 50 years, European countries have introduced newborn bloodspot screening (NBS) for a range of inherited diseases as an important public health program. In 2004, the European CF Society established the Neonatal Screening Working Group (NSWG) to support the implementation of CF NBS program across Europe [4, 5]. CF NBS is the optimal method to diagnose the disease during the asymptomatic period. The primary goal of newborn screening is to decrease morbidity, mortality and associated disabilities. The early diagnosis is associated with improved physical development and slower lung deterioration due to the lower frequency of infections.
All CF NBS programs begin with detection of an elevated immunoreactive trypsinogen (IRT) level in a dried blood specimen from the newborn [3, 5, 6]. A positive IRTscreen is triaged to second-tier testing, which is DNA mutation testing in many national screening programs, including Poland (IRT/DNA protocol) [3,4,5,6,7]. Despite the advent of NBS and improved knowledge about CFTR genetics, CF diagnosis remains incomplete for many reasons, such as inconclusive sweat chloride values, CFTR mutations of uncertain pathogenicity, and differential expression of CFTR or modifier effects [8]. Also CF NBS introduced a new complexity and diagnostic dilemma, namely infants with abnormal screening tests, because of elevated immunoreactive IRT levels but inconclusive sweat tests and/or DNA results. The term for infants with an inconclusive diagnosis have been proposed: CF screen positive, inconclusive diagnosis (CFSPID) in Europe [9].
The first pilot NBS CF study was performed in Poland from 1999 to 2002 and included about 25% of the Polish population. The current program was implemented in 2006 and was expanded to the whole country in the summer of 2009. In Lodz Voivodeship, CF NBS was introduced on 1st June 2009 [3, 6, 7].
The research presents 10 years of experience of our CF center with the national newborn CF screening program in Lodz Voivodship and including the analyzis of diagnostic dilemma occurred during the program. The aim of the study was to determine how early diagnosis of CF, using neonatal screening with IRT/DNA protocol, affects long term clinical outcomes in comparison to CF patients with disease diagnosed before CF NBS?
Material and methods
Patients
Patients participated in the screening program
The study population comprised of infants who were born between 01.07.2009 and 31.12.2019 in Lodz Voivodship and were referred to our CF center as a part of NBS for CF and performed CFTR gene analysis according to IRT/DNA protocol described below. The infants had been evaluated at least once per 3 months. They were divided to three groups according to laboratory tests and further clinical evaluation as CF NBS Group, CF SPID and false positive NBS. Cystic fibrosis (CF NBS Group) was diagnosed in infants based on the level of sweat chloride in addition to evidence of CFTR dysfunction. The designation CF SPID was established to asymptomatic infants if they presented a positive CF NBS test plus: sweat chloride < 59 mmol/L and 2 CFTR mutations with 0–1 CF-causing CFTR mutations. The asymptomatic CF NBS positive infant with presence of no CFTR mutation or mutation in one allele plus a negative sweat test, referred as false positive NBS (including carriers).
Patients diagnosed with CF before introduction of screening program
We also analyzed data of 52 patients (24 females and 28 males) with established CF diagnosis born between 1996 and 2009 (before the newborn screening in Poland) attending our Cystic Fibrosis Outpatient Center in Copernicus Hospital in Lodz, Poland. Patients in this group had been referred to the Center due to occurrence of symptoms suggesting CF (CF Group).
Next, we compared both groups CF NBS and CF. We have chosen the interval — between 5 and 8 years of age to compare both groups, because at that time the most data were available.
Data assessment
In both groups the data were obtained retrospectively after a review of patient charts. The socio-economic data were analysed. These included a place of living defined as a large city above 100,000 inhabitants, a small city 10,000–100,000 inhabitants, and a village below 10,000 inhabitants. The perinatal period contained data: delivery data and weight, Apgar score and presence of meconium ileus (MI), time of CF diagnosis. We also analysed body weight and percentile of body weight at 5 and 8 years of age.
Clinical evaluation
Respiratory status was assessed by the forced expiratory volume in 1 s (FEV1) in spirometry, expressed as percentages of predicted values for height, weight, age, and gender. All analyzed measurements were done in fifth and 8 year of life, only in stable periods (using a Jaeger MasterScreen Body spirometer; E Jaeger GmbH; Wurzburg, Germany). The tests were performed according to American Thoracic Society standards [10].
The number of bronchopulmonary exacerbations and number of hospitalizations (due to pulmonary disease, longer than 3 days) between five and 8 years of life were evaluated. Pulmonary exacerbation was defined as excessive sputum expectoration, general malaise, and need for antibiotics. Chronic bacteria colonization Pseudomonas aeruginosa (PA) or methicillin-resistant Staphylococcus aureus (MRSA) were also collected, and defined as three positive consecutive sputum cultures over a period of 6 months in fifth and 8 year of life.
CF NBS protocol
IRT was measured in dried blood samples from 3 to 5 days old neonates. The IRT concentration cut-off was established as > 99.4 percentile (according to the pilot CF NBS program [6]). The elevated IRT caused further genetic testing from the same dried blood samples. In neonates with meconium ileus (MI), the DNA analysis was performed regardless of IRT level. The measurements of IRT (by using IRT Neonatal Screening ELISA colorimetric assay — IBL International) and DNA analysis from sampling paper (processing with the Extract Blood PCR Kit) were performed in the Genetic Department, Institute of Mother and Child, Warsaw, Poland. Since September 2011, the extended DNA analysis panel has been used and comprised 95% of mutated alleles in the Polish population.
Neonates with one or two mutations in CFTR detected due to NBS examinations were directed to CF Centres for clinical assessment and sweat tests (measured by the quantitative pilocarpine iontophoresis method and Nanoduct method parallel) [6]. CF NBS was conducted as a pilot programme in four Polish districts in the period 1999–2003. In 2006 CF NBS started again in the same regions and was gradually extended across the country. In June 2009 covered all of newborn population in Poland. In the 1980s, the prevalence of CF for Caucasians was estimated at 1: 2500 live births. The disease incidence was more accurately calculated to 1: 4000–5000 thanks to the extensive implementation of CF NBS in the world. In Poland due to data from CF NBS in the years 2006–2010 it is believed that one child every 4394 live births is born with CF.
The study was approved by the Ethical Committee of the Medical University of Lodz, Poland (nr RNN/145/20/KE).
Statistical analysis
Numerical traits were described by way of the arithmetic mean, standard deviation (SD), standard error (SE), when applicable, and their minimum-to-maximum values. Categorical variables were depicted by using counts (n) and percentages (%).
Multiple logistic (for binary dependent variables), multiple linear (for numerical traits), and multiple Poisson (for count data) regression models were fitted in order to test statistical co-dependencies. When dealing with non-normally distributed variables, robust standard errors (i.e. sandwich estimators) were used in the regression equations. All the regression models were controlled for the studied patients’ characteristics such as sex, gender, place of residence, birth weight, APGAR and also for crucial infections (PA, MRSA) before their 5th year of age. Missing data were case-wise deleted.
A level of P < 0.05 was deemed statistically significant. All the computations were performed by using of Stata/Special Edition, release 14.2 (StataCorp LP, College Station, Texas, USA).
Results
Evaluation of patients participated in the screening program (CF NBS positive infants)
One hundred fifty one infants were referred to our CF center as a part of NBS for CF due to CF suspicion: 84 females and 67 males. The CFTR gene analysis for each positive IRT, and neonates with MI regardless of IRT levels, allowed to identify mutations in one allele in 145 patients and in both alleles in 44 patients (96.03% of screened CF patients). Among 44 cases, after biochemical, genetic and clinical evaluations, in 37 children the diagnosis of CF was established (24.5%) during the first verifying visit (CF NBS Group) — in 76% the diagnosis was establish up to 35 days after birth, in 9 patients the first verifying visit took place later in life due to hospitalization connected with their MI. The mean birth weight in CF NBS patients was 3226.34 g, In this group, 9 patients had MI (24.32%). In the remaining 7 (out of 44) cases (15.9%), despite detection of two CFTR mutations, a clinical evaluation did not confirm the CF diagnosis (CF SPID). These infants had the CFTR genotype as follows: 3 patients with [F508del]; [IVS8-5 T+(TG)11], [3849 + 10kbC > T];[R117H], [Y301C];[3271 + 18C > T], [V754M];[V562L], [F508del];[R117H]. The above mutations found at least in one allele causing unproven or uncertain clinical consequences. These patients remain under the observation of our clinic. The most common mutation was F508 del, detected in 104 patients in one or both alleles (68.88%).
Six patients (3.97%) who were directed to the Cystic Fibrosis Outpatient based on the IRT elevations, had DNA analysis for CF mutations negative and they sweat chloride test was correct (false positive NBS). After further observations and examinations, the CF was excluded. So far (until December 2019) we also have information about two cases of false-negative results in CF NBS in our region (false negative NBS).
Comparison of group CF NBS and group CF
At the time of data collection 27 of CF NBS patients were at least 8 years old, remain under the medical care in our CF Outpatient Center and were included into compared analysis. The Group CF NBS with positive result of newborn CF screening included: 59% females, mean birth weight 3168.52 g and mean Apgar 8.78. Mutation F508 del was detected in both alleles in 29.63% patients.
Fifty two patients belonging to the Group CF, were born between 1996 and 2009 (before the start of newborn screening in Poland) – 24 females and 28 males. They were included into compared analysis with Group CF NBS. Their mean birth weight was 3195.6 g, Apgar 8.54, F508 del mutation in both alleles was detected in 50% cases.
Group CF NBS and Group CF demographic and clinical characteristics are shown in Table 1.
Full size table
The average time of the CF diagnosis was 45.25 months from birth in Group CF.
and 1.59 months in Group CF NBS.
The most significant difference between this two Groups was the number of exacerbations and hospitalizations between 5 and 8 years of life (p < 0.001). Pulmonary exacerbation in analysed period occurred on average 4.2 times in children in Group CF NBS and they were hospitalized on average 0.5 times. While in Group CF children mean of pulmonary exacerbations was 6.77 and mean number of hospitalization was 2.14.
Even though the average body weight and its percentile value was higher in the Group CF NBS, this difference was not significant. What is more weight gain between 5 and 8 years old was greater in the Group CF. No statistically significant predictors affecting weight gain in both groups were defined.
In spirometry measurements patients in both groups presented stable values of FEV1 during at least 3 years of observation, and the difference was not significant.
The number of PA infected patients increased significantly between the fifth and eighth year of age of the examined children (OR = 1.16 (95% CI: 1.04–19) (P = 0.007)) regardless of the study group (P = 0.984). It seems that patients with MRSA infection in the fifth year of life have a higher risk of PA infections in subsequent years of their life (OR = 1.45 (95% CI: 1.03–2.03) (P = 0.032)). The number of patients with PA or MRSA infection in the fifth year of life and PA infection in the age of 8 was greater in the CF Group, but these differences were not statistically significant.
Descriptive characteristics of all CF patients by screening status are shown in Table 2.
Full size table
Discussion
According to the Neonatal Screening Working Group new-born screening for CF provides an immediate diagnosis, before the onset of clinical symptoms [11,12,13]. In the beginning CF NBS raised doubts about ethical aspects with regard to possible benefits and risks. After many years of experience CF NBS has been widely implemented and accepted. Ten years of CF NBS programme in Lodz Voivodship leads to diagnosis of CF in 37 neonates among 151 children directed after their birth with CF suspicion to our Outpatient Center. The incidence of CF in Poland based on neonatal screening is currently 1:4394–1:5000 [3, 6].
CF NBS has been based on the assumption that pre-symptomatic detection permits early access to specialised medical care, and thus results in less morbidity and longer life expectancy. In our region CF NBS screened patients were diagnosed and treated in multidisciplinary CF centre between first and second month of their life. For comparison the median age of the diagnosis of patients, who were born before the start of CF NBS in Poland due to typical CF symptoms – 45.25 months from their birth. According to the literature, the most benefits associated with early identification of CF including include better growth and lung function, less intensive therapeutic burden and reduced cost of care [11,12,13,14]. Our observations show that early diagnosis and the introduction of appropriate treatment for asymptomatic CF patients resulted in a lower frequency of the pulmonary exacerbation and lower number of hospitalizations. It is very beneficial observation due to reports which emphasise that the repeated mild-to-moderate pulmonary exacerbations, especially in the first years of life result in airway remodelling. While more severe hospitalised episodes could increase structural airway injury risk [13,14,15,16]. Data from Australia showed improved survival in future in patients with lower number of hospitalisations in the first 3 years of life [17]. The occurrence of respiratory exacerbations are associated with an accelerated decline in lung function and reduced quality of life and survival [15,16,17]. On the other hand mean values of FEV1 in the analysed patients, presented stable results during at least 3 years of observation; there was no difference between Groups. This may be due to the age — below 8 — too early to identify significant differences in FEV1 secondary to clinical outcome (such as pulmonary exacerbations, bacterial colonisations), which likely takes longer to cause impairment of lung function that is identifiable with spirometry [16].
Children with CF are highly susceptible to chronic respiratory tract colonization and subsequent recurrent infection. It is know that once chronic PA infection is established, the risk of mortality and morbidity increases [18, 19]. In our study the mean number of patients with PA or MRSA colonization in the 5 years old and PA chronic infection in the age of 8 was lower among children screened in NBS, but there were no significant differences between both groups. The number of PA infected persons increased significantly between the fifth and eighth year of age regardless of belonging to the study group. Although the increasing number of SA and PA colonized patients were observed with age in both CF NBS and CF group, the CF NBS children had fewer exacerbations and hospitalisations due to intensively treatment from the very first PA sputum detection. The important observation was that patients in both groups with SA infection in the fifth year of life have a higher risk of PA infections in subsequent years of their life. Persistent presence of MRSA in the respiratory tract is associated with increased rates of PA through mediation of PA biofilm formation [18, 19]. In practice, we suggest to employ intermittent, symptom-based treatment of SA especially MRSA to delay initial PA colonization. The colonization with PA remains the most ordinary airway pathogen contributing to shortened survival in CF patients. For over a two decades antibiotic effective for PA (for example tobramycin inhalation solution (TIS)) is recommended for the treatment and early introduction with TIS has resulted in improved lung function and reduced risk of airway exacerbations [20, 21]. In our study in CF group the diagnosis was established mean in 42.25 month of life, but even maximally in 180 months of life. In such situation at the time of CF recognition patients’ sputum were often positive for PA and we treated PA colonization not an initial infection. Patients from CF NBS group were treated intensively from the very first PA sputum detection. We believe this may have contributed to a reduction in the number of pulmonary exacerbations and associated hospitalizations. Moss et al. [21] proved that delay in initiation of TIS therapy is associated with reduced long-term improvement over baseline, suggesting an irreversible component to lung function decline. To be maximally effective, TIS treatment should begin early after first detection of PA. In agreement to this study, although the incidence of PA found in the airways became similar over the years in both study groups (CF and CF NBS) the number of exacerbations and hospitalizations in CF NBS group was significant lower.
CF NBS allows the affected infants to receive immediate treatments including not only physiotherapy and preventing of pulmonary manifestations of the disease, but also great emphasis is pleased on assessment of nutritional status, and pancreatic enzyme replacement therapy. The Wisconsin randomised trial on newborn screening for CF showed that screened neonates exhibit better nutritional status in the first years of life [22], but no output is available from this cohort on long-term survival. We observed trend that body weight were higher in the CF NBS Group and they presented systematic and stable weight gain but this results were not statistically significant.
According to data published by Bobadilla et al., the complete CFTR gene analysis by sequencing of selected regions should identify mutations in one or both alleles in nearly 95% of screened children [23] and IRT/DNA strategy to achieve sensitivity close to 100% (despite children with MI, whose IRT level is not elevated). Due to Fritz formula the predicted false-negative rate for the Polish population is 6–7 cases per year, which gives 1–2 omitted CF cases per 100,000 live births [24]. It shows that despite the high sensitivity and specificity of IRT/DNA protocol it is important to follow patients with CF suggestive symptoms even when CF NBS was negative. We had two cases of false-negative results in CF NBS in our region to this day. According to normal values of the IRT in new-borns with MI they are in risk group of a false-negative first step of CF NBS protocol. Each case of MI should be reported to the NBS center by appropriate note on sampling paper and neonates must to be verify in CF centers. In our study, the new-borns with MI represent 24% of CF diagnosis, while the literature data shows MI frequency about 10–20% [25].
The IRT/DNA strategy allows to detect not only CF disease but also carries of the CFTR gene mutations, as well as cases of unclear clinical consequences. This fact constitutes a definite psychological problem for patients and their parents and raised doubts about ethical aspects of the CF NBS. It may have negative influences on further procreation of such persons. We detected 101 patients with mutations only in one allele of CFTR gene (what was 66.89% of all neonates from CF NBS). They were defined as carriers and in all cases, a genetic consultations for their parents and siblings was recommended.
Some limitations of our current study include a single centre analysis, which limits its generalizability. What is more we made the comparison to historical data (a blinded controlled study with CF- newborns left untreated was unacceptable due to ethical reasons).
Over the years new therapies were approved, standards of care established therefore, the outcomes of same patients in subsequent years is expected to be improved, even without the advent of newborn screening. We have to choose the appropriate time interval to compere a historical cohort (CF Group) and CF NBS positive patients.
Conclusion
In conclusion, newborn screening contributes to early diagnosis of cystic fibrosis. The presented study showed that CF NBS has beneficial effects primarily on decrease of pulmonary exacerbations with hope for a longer life expectancy in these group.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CF:
-
Cystic fibrosis
- CFNBS:
-
Cystic Fibrosis newborn screening
- CFTR:
-
CF transmembrane conductance regulator
- FEV1:
-
The forced expiratory volume in 1 s
- IRT:
-
Immunoreactive trypsinogen
- MI:
-
Meconium ileus
- NSWG:
-
Neonatal Screening Working Group
- PA:
-
Pseudomonas aeruginosa
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- TIS:
-
Tobramycin inhalation solution
References
-
Farrell PM. The prevalence of cystic fibrosis in the European Union. J Cyst Fibros. 2008;7(5):450–3. https://doi.org/10.1016/j.jcf.2008.03.007.
Article
PubMedGoogle Scholar
-
Paranjapye A, Ruffin M, Harris A, Corvol H. Genetic variation in CFTR and modifier loci may modulate cystic fibrosis disease severity. J Cyst Fibros. 2019;13(19):30963–4.
Google Scholar
-
Sobczyńska-Tomaszewska A, Ołtarzewski M, Czerska K, Wertheim-Tysarowska K, Sands D, Walkowiak J, et al. Newborn screening for cystic fibrosis: polish 4 years experience with CFTR sequencing strategy. European J Human Genet. 2013;21(4):391–6. https://doi.org/10.1038/ejhg.2012.180.
Article
CASGoogle Scholar
-
Barben J, Castellani C, Dankert-Roelse J, Gartner S, Kashirskaya N, Linnane B, et al. The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe. J Cyst Fibros. 2017;16(2):207–13. https://doi.org/10.1016/j.jcf.2016.12.012.
Article
PubMedGoogle Scholar
-
Loeber JG, Burgard P, Cornel MC, Rigter T, Weinreich SS, Rupp K, et al. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 1. From blood spot to screening result. J Inherit Metab Dis. 2012;35(4):603–11. https://doi.org/10.1007/s10545-012-9483-0.
Article
PubMedGoogle Scholar
-
Sands D, Zybert K, Mierzejewska E, Ołtarzewski M. Diagnosing cystic fibrosis in newborn screening in Poland — 15 years of experience. Dev Period Med. 2015;19(1):16–24.
PubMed
Google Scholar
-
Zietkiewicz E, Rutkiewicz E, Pogorzelski A, Klimek B, Voelkel K, Witt M. CFTR mutations spectrum and the efficiency of molecular diagnostics in polish cystic fibrosis patients. PLoS One. 2014;26(2):8909.
Google Scholar
-
Levy H, Nugent M, Schneck K, Stachiw-Hietpas D, Laxova A, Lakser O, et al. Refining the continuum of CFTR-associated disorders in the era of newborn screening. Clin Genet. 2016;89(5):539–49. https://doi.org/10.1111/cge.12711.
Article
CAS
PubMed
PubMed CentralGoogle Scholar
-
Ren C, Borowitz D, Gonska T, Howenstine M, Levy H, Massie J, et al. Cystic fibrosis transmembrane conductance regulator-related metabolic syndrome and cystic fibrosis screen positive, inconclusive diagnosis. J Pediatr. 2017;181:45–51.
Article
Google Scholar
-
Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi A, Coates A, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–38. https://doi.org/10.1183/09031936.05.00034805.
Article
CASGoogle Scholar
-
Castellani C, Massie J, Sontag M, Southern KW. Newborn screening for cystic fibrosis. Lancet Respir Med. 2016;4(8):653–61. https://doi.org/10.1016/S2213-2600(16)00053-9.
Article
PubMedGoogle Scholar
-
Tridello G, Castellani C, Meneghelli I, Tamanini A, Assael BM. Early diagnosis from newborn screening maximises survival in severe cystic fibrosis. ERJ Open Res. 2018;4:2.
Article
Google Scholar
-
Castellani C, Linnane B, Pranke I, Cresta F, Sermet-Gaudelus I, Peckham D. Cystic fibrosis diagnosis in newborns, children, and adults. Semin Respir Crit Care Med. 2019;40(6):701–14. https://doi.org/10.1055/s-0039-1697961.
Article
PubMedGoogle Scholar
-
Barben J, Castellani C, Dankert-Roelse J, Gartner S, Kashirskaya N, Sands D. Et all. The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe. J Cyst Fibros. 2017;16(2):207–13. https://doi.org/10.1016/j.jcf.2016.12.012.
Article
PubMedGoogle Scholar
-
Byrnes C, Vidmar S, Cheney J, Carlin J, Armstrong D, Cooper P, et al. Prospective evaluation of respiratory exacerbations in children with cystic fibrosis from newborn screening to 5 years of age. Thorax. 2013;68(7):643–51. https://doi.org/10.1136/thoraxjnl-2012-202342.
Article
PubMed
PubMed CentralGoogle Scholar
-
Goss C. Acute pulmonary exacerbations in cystic fibrosis. Semin Respir Crit Care Med. 2019;40(6):792–803. https://doi.org/10.1055/s-0039-1697975.
Article
PubMed
PubMed CentralGoogle Scholar
-
Reid DW, Blizzard CL, Shugg DM, Flowers C, Cash C, Greville HM. Changes in cystic fibrosis mortality in Australia, 1979-2005. Med J Aust. 2011;195(7):392–5. https://doi.org/10.5694/mja10.11229.
Article
PubMedGoogle Scholar
-
Mostofian F, Alkadri J, Tang K, Thampi N, Radhakrishnan D. A real world evaluation of the long-term efficacy of strategies to prevent chronic Pseudomonas aeruginosa pulmonary infection in children with cystic fibrosis. Int J Infect Dis. 2019;85:92–7. https://doi.org/10.1016/j.ijid.2019.05.026.
Article
PubMedGoogle Scholar
-
Armbruster CR, Wolter DJ, Mishra M, Hayden HS, Radey MC, Merrihew G, et al. Staphylococcus aureus protein A mediates interspecies interactions at the cell surface of Pseudomonas aeruginosa. MBio. 2016;7:3.
Article
Google Scholar
-
Sawicki GS, Signorovitch JE, Zhang J, Latremouille-Viau D, vonWartburg M, Wu EQ, et al. Reduced mortality in cystic fibrosis patients treated with tobramycin inhalation solution. Pediatr Pulmonol. 2012;47(1):44–52. https://doi.org/10.1002/ppul.21521.
Article
PubMedGoogle Scholar
-
Moss RB. Long-term benefits of inhaled tobramycin in adolescent patients with cystic fibrosis. Chest. 2002;121(1):55–63. https://doi.org/10.1378/chest.121.1.55.
Article
CAS
PubMedGoogle Scholar
-
Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin cystic fibrosis neonatal screening study group. Pediatrics. 2001;107(1):1–13. https://doi.org/10.1542/peds.107.1.1.
Article
CAS
PubMedGoogle Scholar
-
Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations – correlation with incidence data and application to screening. Hum Mutat. 2002;19(6):575–606. https://doi.org/10.1002/humu.10041.
Article
CAS
PubMedGoogle Scholar
-
Fritz A, Farrell P. Estimating the annual number of false negative cystic fibrosis newborn screening tests. Pediatr Pulmonol. 2011;47(2):207–8. https://doi.org/10.1002/ppul.21561.
Article
PubMedGoogle Scholar
-
Tan SMJ, Coffey MJ, Ooi CY. Differences in clinical outcomes of paediatric cystic fibrosis patients with and without meconium ileus. J Cyst Fibros. 2019;18(6):857–62. https://doi.org/10.1016/j.jcf.2019.09.008.
Article
PubMedGoogle Scholar
Download references
Acknowledgements
Not applicable.
Author information
Authors and Affiliations
-
Department of Pediatrics and Allergy, Medical University of Lodz, Copernicus Memorial Hospital, Korczak Paediatric Center, Piłsudskiego 71 Str, 90-329, Lodz, Poland
M. Olszowiec-Chlebna, E. Mospinek & J. Jerzynska
Authors
- M. Olszowiec-Chlebna
You can also search for this author in
PubMed Google Scholar - E. Mospinek
You can also search for this author in
PubMed Google Scholar - J. Jerzynska
You can also search for this author in
PubMed Google Scholar
Contributions
M. Olszowiec-Chlebna MD PhD (literature search, study design, analysis of data, manuscript preparation), E. Mospinek MD (literature search, analysis of data). J. Jerzynska MD, PhD, Prof (study design, manuscript preparation, review of manuscript). The author(s) read and approved the final manuscript.
Corresponding author
Correspondence to
J. Jerzynska.
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethical Committee of the Medical University of Lodz, Poland (nr RNN/145/20/KE).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and Permissions
About this article
Cite this article
Olszowiec-Chlebna, M., Mospinek, E. & Jerzynska, J. Impact of newborn screening for cystic fibrosis on clinical outcomes of pediatric patients: 10 years’ experience in Lodz Voivodship.
Ital J Pediatr 47, 87 (2021). https://doi.org/10.1186/s13052-021-01040-5
Download citation
-
Received: 03 November 2020
-
Accepted: 26 March 2021
-
Published: 09 April 2021
-
DOI: https://doi.org/10.1186/s13052-021-01040-5
Keywords
- Cystic fibrosis
- New born screening
- Diagnosis
- IRT/DNA protocol
- Children
Актуальные вопросы диагностики муковисцидоза
Статьи
![]()
ЖУРНАЛ «ПРАКТИКА ПЕДИАТРА»
Опубликовано в журнале:
«ПРАКТИКА ПЕДИАТРА»; март-аперль; 2015; стр. 20-27.
Е.И. Кондратьева, д. м. н., профессор, В.Д. Шерман, к. м. н., Н.И. Капранов, д. м. н., профессор, Н.Ю. Каширская, д. м. н., профессор, НКО муковисцидоза ФГБНУ «МГНЦ», ГБУЗ «ДГКБ № 13 им. Н.Ф. Филатова ДЗМ», г. Москва
Муковисцидоз (МВ), или кистозный фиброз (cysticfibrosis), — одно из наиболее частых моногенных наследственных заболеваний с полиорганной патологией, резко сокращающее продолжительность и качество жизни пациентов без адекватного комплексного лечения в течение всей жизни. МВ распространен среди населения всей Земли, но наиболее часто поражает европеоидов: в среднем с частотой 1 на 2500-4500 новорожденных. Еще совсем недавно больные муковисцидозом умирали в раннем детском возрасте или даже на первом году жизни от пневмонии и истощения, обусловленными мальабсорбцией.
Ключевые слова: диагностика, генетика, мутации, неонатальный скрининг, потовая проба, эластаза кала.
Key words: cystic fibrosis, diagnosis, genetics, mutation, newborn screening, sweat test, fecal elastase.
Болезнь прежде всего характеризуется повышенной продукцией вязкого бронхиального секрета, частыми легочными инфекциями и обструкцией дыхательных путей. По мере прогрессирования легочной болезни образуются участки ателектазов, развивается эмфизема, постепенно разрушается паренхима легких с развитием бронхоэктазов и участков пневмосклероза, а больной имеет высокий риск погибнуть от легочно-сердечной недостаточности. В финальной стадии заболевания пересадка комплекса «сердце-легкие» остается для больного единственной надеждой. Помимо бронхолегочной системы у большинства больных муковисцидозом поражается поджелудочная железа, при этом это происходит внутриутробно. Недостаточность панкреатических ферментов обусловливает нарушение всасывания жиров и белков, развитие нутритивной недостаточности. В результате больные отстают в росте и страдают гипотрофией. Продукция инсулина также может быть нарушена, что ведет к развитию диабета. К частым осложнениям течения муковисцидоза относят остеопороз, а также жировой гепатоз с переходом в цирроз. При наличии «мягкой» мутации клинические проявления развиваются постепенно, преобладают моносимптомы, диагноз «муковисцидоз» устанавливается поздно или случайно.
Своевременная диагностика муковисцидоза, обеспечивающая в большинстве случаев раннее начало терапии, в том числе на доклиническом этапе, улучшает прогноз заболевания, повышает эффективность лечения, позволяет предупредить развитие тяжелых осложнений, значительного отставания в физическом развитии, а в ряде случаев и необратимых изменений в легких. Ранняя диагностика позволяет семье вовремя решить необходимые вопросы, связанные с рождением здорового ребенка (генетическое консультирование, пренатальная диагностика МВ в последующие беременности).
Диагностика делится на:
1) пренатальную диагностику;
2) диагностику по неонатальному скринингу (до клинических проявлений или при их дебюте);
3) диагностику при клинических проявлениях:
4) диагностику среди родственников больных.
В настоящее время налаживается дородовая диагностика муковисцидоза в перспективных и информативных семьях (Москва, Санкт-Петербург, Уфа, Томск, Красноярск, Ростов-на-Дону, Владивосток и некоторые другие города), что, безусловно, важно для профилактики этой тяжелой патологии. Пренатальная диагностика возможна в виде ДНК-диагностики при проведении амниоцентеза (получение околоплодных вод в ранний срок -13-14 недель и поздний — обычно 16-20 недель беременности) в семье носителей одной мутации гена CFTR и имеющей больного ребенка. Диагноз может быть заподозрен при УЗИ плода внутриутробно при наличии характерной УЗ-характеристики в виде гиперэхогенного кишечника. УЗИ во время беременности рекомендуют в скрининговые сроки: 11-14, 18-21 и 30-34 недели беременности. Обязательно проводят повторное исследование. В 50-78% случаев это состояние будет связано с МВ и проявится мекониальным илеусом. Диагноз в этом случае может быть установлен еще до рождения ребенка. В то же время этот признак не является высокоспецифичным для МВ, может быть транзиторным явлением, а также связанным с другими патологическими состояниями. При этом ДНК-диагностика родителей дает необходимую информацию о наличии мутаций у каждого из родителей и позволяет предполагать заболевание у ребенка при рождении.
Клинические признаки
1. Диагностика классической формы МВ обычно не представляет сложностей. Классический фенотип больного является результатом наличия двух мутантных копий гена муковисцидозного трансмембранного регулятора (CFTR) и характеризуется хронической бактериальной инфекцией дыхательных путей и придаточных пазух носа, стеатореей из-за внешнесекреторной недостаточности поджелудочной железы, мужским бесплодием из-за обструктивной азооспермии, а также повышенной концентрацией хлоридов потовой жидкости.
2. Проблемы диагностики МВ, как правило, связаны с фенотипическим разнообразием его форм, обусловленным генетическим полимор-
В ряде случаев атипичного течения МВ возможна его диагностика во взрослом возрасте. Как правило, в этой группе больных отмечается более мягкое течение болезни в связи с сохранностью функции поджелудочной железы и нетяжелым поражением органов дыхания.
В абсолютном большинстве случаев МВ может быть диагностирован в раннем детском возрасте (в 90% случаев — на первом году жизни). К сожалению, нередки случаи диагностики МВ у взрослых с классическим фенотипом.
Диагностика МВ у носителей «мягких» генотипов (актуально для детей, рожденных до 2006-2007 гг., и взрослых):
В настоящее время выделяют несколько групп риска по МВ.
Основной группой риска по заболеванию в РФ в настоящее время являются новорожденные с неонатальной гипертрипсиногенемией. Учитывая возможность получения ложноотрицательных результатов неонатального скрининга, а также то обстоятельство, что в РФ неонатальный скрининг на МВ проводится с 2006-2007 гг., не теряет своей актуальности анализ групп риска, включающих пациентов с патологией желудочно-кишечного тракта, бронхолегочными нарушениями, патологией других органов и родственников больных МВ (табл. 1).
Таблица 1.
Группы риска для дифференциальной диагностики муковисцидоза
| I. Бронхолегочные нарушения |
| 1. Повторные и рецидивирующие пневмонии с затяжным течением, особенно двусторонние 2. Бронхиальная астма, рефрактерная к традиционной терапии 3. Рецидивирующие бронхиты, бронхиолиты, особенно с высевом Ps. aeruginosa 4. Двусторонние бронхоэктазы |
| II. Изменения со стороны желудочно-кишечного тракта |
| 1. Синдром нарушенного кишечного всасывания неясного генеза 2. Мекониальный илеус и его эквиваленты 3. Гиперэхогенность кишечника плода 4. Желтуха обструктивного типа у новорожденных с затяжным течением 5. Цирроз печени 6. Сахарный диабет 7. Гастроэзофагеальный рефлюкс 8. Выпадение прямой кишки |
| III. Патология со стороны других органов |
| 1. Нарушение роста и развития 2. Задержка полового развития 3. Мужское бесплодие 4. Хронический синусит 5. Полипы носа 6. Электролитные нарушения |
| IV. Члены семей больных муковисцидозом |
Среди клинических проявлений, характерных для МВ, можно выделить высоко-и менее специфичные (табл. 2). Состояния, представленные в левой колонке таблицы, в абсолютном большинстве случаев встречаются у больных МВ. Причиной состояний из правой колонки могут быть другие заболевания, например первичная цилиарная дискинезия, гуморальный иммунодефицит и т. д.
Таблица 2.
Клинические проявления, характерные для МВ
| Высокоспецифичные для МВ | Менее специфичные для МВ |
| Желудочно-кишечные:
|
Желудочно-кишечные:
|
| Со стороны дыхательных путей:
|
Со стороны дыхательных путей:
|
| Другое:
|
Другое:
|
В таблице 3 представлены особенности проявлений МВ в разные возрастные периоды. Знание этих особенностей помогает специалистам, наблюдающим пациента с теми или иными симптомами, включить МВ в перечень заболеваний для дифференциальной диагностики. Особенно это касается детей раннего возраста, когда клиническая картина еще может быть неполной, но на себя будут обращать внимание некоторые проявления, например мекониальный илеус при рождении или синдром потери солей, не имеющий связи с патологией почек. Диагноз в этом случае может быть установлен еще до рождения ребенка. В то же время этот признак не является высоко специфичным для МВ, может быть транзиторным явлением, а также связанным с другими патологическими состояниями.
Таблица 3.
Клинические особенности проявлений МВ в различные возрастные периоды
| 0-2 года | |
|
|
|
| 3-16 лет | |
|
|
|
Диагностические критерии МВ
Для решения проблем диагностики МВ, в том числе и его атипичных форм, были разработаны критерии, согласно которым обязательным для МВ является наличие характерного клинического синдрома плюс доказательство какого-либо нарушения функции хлорного канала.
Учитывая все научные достижения в понимании природы муковисцидоза и МВ-зависимых заболеваний за последние 10 лет, в 2013 году группа экспертов Европейского общества муковисцидоза (European Cystic Fibrosis Society) под руководством Carlo Castellani подготовила новые стандарты диагностики в редакции Alan R. Smyth и Scott Bell (схема).
Схема.
Диагностические критерии муковисцидоза ECFS 2013
| Положительная потовая проба и/или две мутации МВТР, вызывающие МВ (согласно базе CFTR-2) |
И | Неонатальная гипертрипсиногенемия или характерные клинические проявления, такие как диффузные бронхоэктазы, высев из мокроты значимой для МВ патогенной микрофлоры (особенно синегнойной палочки), экзокринная панкреатическая недостаточность, синдром потери солей, обструктивная азооспермия |
Неонатальный скрининг
Проводится на основании Методических рекомендаций по проведению неонатального скрининга в РФ с использованием Европейских рекомендаций по неонатальному скринингу. 90% новорожденных без клинических проявлений муковисцидоза диагноз может быть установлен на основании скрининга в возрасте до 6 недель. В 5-10% случаев возникают трудности с диагностикой муковисцидоза (Cystic Fibrosis Foundation Patient Registry, 2005 Annual Data Report to the Center Directors. Bethesda, MD: CFF).
Проблемы неонатального скрининга:
Потовая проба
Показания:
1. При положительном результате неонатального скрининга (двукратном повышении уровня иммунореактивного трипсиногена в крови в течение первого месяца жизни ребенка).
2. При наличии у пациента каких-либо характерных клинических проявлений МВ.
3. Случаи МВ в семье.
Потовая проба является надежным методом диагностики МВ у 98% больных. Исследование можно проводить всем детям через 48 часов после рождения, хотя у новорожденных могут быть проблемы с набором пота. Несмотря на то, что «золотым стандартом» диагностики МВ считается количественное определение хлоридов в потовой жидкости (классический метод Гибсона — Кука), метод определения проводимости на аппаратах «Макродакт» и «Нанодакт» («Вескор», США) показал хорошую с ним корреляцию в многочисленных исследованиях.
Оценка результата
При положительном результате потовой пробы (хлориды > 60 ммоль/л при классическом методе Гибсона — Кука и/или проводимость > 80 ммоль/л NaCl) диагноз подтверждается.
Генетическое исследование
Генетическое исследование проводится после потовой пробы. Однако в связи с ограниченными возможностями ДНК-диагностики в России данный метод не является обязательным, однако применяется с исследовательской целью и для окончательного подтверждения диагноза.
На первом этапе ДНК-обследования наиболее часто используется панель, включающая 28 мутаций, как наиболее частых в мире, так и специфичных для России: F508del, CFTRdele2,3(21kb), 3849+10kbC>T, W1282X, 2143delT, 2184insA, 1677delTA, N1303K, G542X, R334W, E92K, L138ins, 394delTT, 3821delT, S1196X, 2789+5G>A, G85E, 2183AA>G, 604insA, 621+1G>T, R117H, R347P, R553X, 3667insTCAA, G551D, I507del, 1717-1G>A, 2184delA. По данным лаборатории генетической эпидемиологии ФГБУ «Медико-генетический научный центр» (МГНЦ) РАМН, при использовании данной панели удается обнаружить лишь около 82,5% мутантных аллелей у больных МВ. В случае когда при положительной потовой пробе не будет найдено ни одной мутации гена (что само по себе маловероятно), может потребоваться секвенирование гена МВ, позволяющее идентифицировать примерно 98% мутаций в гене CFTR.
Рекомендации:
1. На основании данных национального регистра больных МВ по ДНК-диагностике гена CFTR установлены особенности характера и частоты мутаций в регионах страны. На основе данных регистра рекомендуется создание региональных рекомендаций по определению мутаций со ссылкой на регистр (последнюю версию).
2. Отсутствие мутациий без проведения секвенирования — недостаточно для исключения МВ.
3. Некоторые мутации МВТР (3849+10 kb C>T) ассоциированы с нормальным или пограничным результатом потового теста.
4. «Мягкие» мутации характеризуются поздним дебютом заболевания, пограничным значением потовых проб, выявляются чаще при секвенировании.
5. Пациенты с пограничными результатами потовых проб (хлориды 30-60 ммоль/л и/или проводимость 50-80 ммоль/л), единственной мутацией гена представляют реальные трудности для диагностики.
Для диагностики МВ или его исключения при пограничных результатах пробы необходимо:
В европейских странах для подтверждения дефекта ионного транспорта применяется метод определения разности назальных потенциалов или измерение электрического тока в биоптате кишки, отражающие нарушение функции хлорного канала. Оба метода основаны на электрическом характере транспорта ионов и являются высокоинформативными для диагностики МВ.
Диагностика панкреатической недостаточности включает:
У больных МВ показатель эластазы может снижаться в течение первых лет жизни, поэтому определяется в динамике. Низкий уровень панкреатической эластазы расценивается как один из признаков МВ. Приблизительно 1% пациентов с МВ имеет пограничный результат потового теста в комплексе с сохранной функцией поджелудочной железы и хроническим бронхитом.
Диагностика хронического бронхолегочного процесса:
В качестве дополнительных диагностических маркеров могут быть использованы азооспермия в постпубертатном возрасте, идентификация МВ-ассоциированных патогенов из респираторного тракта, рентгенологические признаки синусита.
Знание основных симптомов МВ и особенностей его течения в разные возрастные периоды позволяет своевременно заподозрить наличие заболевания и направить пациента для дальнейшего обследования. Нередкие случаи поздней диагностики МВ связаны как с отсутствием у врачей достаточных знаний о заболевании, так и с фенотипическим разнообразием его форм. Ограниченные возможности ДНК-диагностики МВ в России и ее низкая доступность затрудняют и затягивают окончательную верификацию заболевания.
ЛИТЕРАТУРА
1. Муковисцидоз. Под ред. Н.И. Капранова, Н.Ю. Каширской. М.: ИД «МЕДПРАКТИКА-М», 2014, 672 с. ISBN 978-5-98803-314-1
2. Welsh M.J., Ramsey B.W., Accurso F.J., Cutting G.R. Cystic fibrosis. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., eds. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill, 2001: 5121-88.
3. European cystic fibrosis society standards of care working group. Best practice guidelines. В редакции Alan R. Smith и Scott Bell, 2014.
4. Farell P.M., Rosenstein B.J., White T.B. et al. Cystic fibrosis foundation. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report // J. Pediatr., 2008; 153 (2): S4-S14.
5. Красовский С.А., Каширская Н.Ю., Усачева М.В., Амелина Е.Л., Черняк А.В., Науменко Ж.К. Влияние возраста постановки диагноза и начала специфической терапии на основные клинико-лабораторные проявления заболевания у больных муковисцидозом // Вопросы современной педиатрии, 2014, т. 13, № 2, с. 36-43.
6. de Boeck K., Wilschanski M., Castellani C. et al. Cystic fibrosis: terminology and diagnostic algorithms. Thorax, 2006; 61: 627-635.
7. de Oronzo M.A. Hyperechogenic fetal bowel: an ultrasonographic marker for adverse fetal and neonatal outcome? // J. Prenat. Med., 2011 Jan-Mar; 5 (1): 9-13.
8. Bombieri C. et al. Recommendations for the classification of diseases as CFTR-related disorders // Journal of Cystic Fibrosis, 2011, vol. 10, suppl. 2; S86-S102.
9. Hall E., Lapworth R. Use of sweat conductivity measurements. Annals of Clinical Biochemistry, 2010; 47: 390-392.
10. Sands D., Oltarzewski M., Nowakowska A., Zybert K. Bilateral sweat tests with two different methods as a part of cystic fibrosis newborn screening (CF NBS) protocol and additional quality control. Folia Histochem Cystobiol., 2010 Sep 30; 48 (3): 358-65.
11. Sezer R.G., Aydemir G., Akcan A.B. et al. Nanoduct sweat conductivity measurements in 2664 patients: relationship to age, arterial blood gas, serum electrolyte profiles and clinical diagnosis // J. Clin. Med. Res., 2013 Feb; 5 (1): 34-41.
12. Петрова Н.В. Молекулярно-генетические и клинико-генотипические особенности муковисцидоза в российских популяциях. Автореф. дисс. докт. биол. наук. М., 2009, 42 с.
13. Derichs N., Sanz J., Von Kanel T. et al. Intestinal current measurement for diagnostic classification of patients with questionable cystic fibrosis: validation and reference data. Thorax, 2010 Jul; 65 (7): 594-9.
14. Servidoni M.F., Sousa M., Vinagre A.M. et al. Rectal forceps biopsy procedure in cystic fibrosis: technical aspects and patients perspective for clinical trials feasibility. BMC Gastroenterol., 2013 May 20; 13 (1): 91.
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
Неонатальный скрининг новорожденных (НС) применяется для ранней диагностики муковисцидоза (МВ), позволяя начать лечение до появления очевидных симптомов. Как правило, этот скрининг включает в себя измерение уровня иммунореактивного трипсиногена (ИРТ) в первую неделю жизни.
После положительных результатов этого теста (ИРТ обычно повышается у пациентов с МВ) новорожденные направляются на подтверждающее тестирование, которое включает измерение количества хлоридов в потовой жидкости и генетическое тестирование на мутации в гене трансмембранного гена муковисцидоза (CFTR).
В некоторых случаях результаты НС недостаточны для подтверждения или опровержения диагноза, особенно в случаях нетипичной клинической картины в сочетании с наличием редких мутаций CFTR.
В новом докладе описывается случай 1-летнего ребенка в Польше, диагноз которого был поставлен с большим трудом.
В двухмесячном возрасте ребенок был госпитализирован с проявлениями инфекции верхних дыхательных путей. Через две недели мальчика снова госпитализировали из-за плохого набора веса и рвоты.
При рождении у ребенка отмечен положительный скрининг, выявлено повышение уровня ИРТ, свидетельствующие о возможном наличии МВ. Однако результаты потового теста у пациента не показало положительных результатов, так из четырех измерений только одно было положительным и указывало на наличие болезни. Первоначальный генетический тест на наличие более 700 мутаций, диагностируемых при МВ, не дал положительного результата.
Ребенок получал антибактериальные и противогрибковые препараты с целью купирования возможной легочной инфекции, хотя видимого клинического эффекта не отмечалось. Пациенту был установлен назогастральный зонд для обеспечения адекватного питания.
Дальнейшие исследования, включая анализ мочи пациента, свидетельствовали о нарушении обмена веществ, но окончательных результатов получено не было. У пациента также была обнаружена анемия и отеки.
Тесты на определение хлоридов в потовой жидкости были вновь повторены с использованием нескольких методик и результаты были положительными, что подтвердило наличие МВ. Дальнейшее метаболическое исследование также исключило ряд болезней со схожей клинической картиной.
Дополнительно к выполненным исследованиям был полностью секвенирован ген CFTR, обнаружены идентичные мутации (получившие название c.4035_4038dupCCTA) в обеих копиях гена (по одной унаследованной от каждого биологического родителя, ни у одного из которых не было симптомов болезни).
После нескольких месяцев комплексного лечения, включая высокобелковую диету и заместительную терапию панкреатическими ферментами, удалось добиться нормализации нутритивного статуса.
В целом, своевременная постановка диагноза была затруднена вследствие совокупности ряда факторов, пишут исследователи. Во-первых, неклассическое течение болезни, которое еще больше осложнилось неоднозначными результатами лабораторных исследований, включая и анализ хлорида в потовой жидкости.
Отрицательные генетические тесты также затрудняли постановку диагноза. Команда исследователей отметила, что первоначальное генетическое тестирование исключило 86% мутантных аллелей гена CFTR в польской популяции.
Более того, обнаруженная мутация является довольно редкой, поскольку ранее сообщалось, что она встречается только у поляков и только в сочетании с другими патогенными мутациями.
Группа пришла к выводу, что “описанный случай демонстрирует необычное течение заболевания”, и предположила, что “клинические симптомы пациента и лабораторные данные в сочетании с результатами молекулярных тестов дают полезную информацию для дальнейшего изучения генотип-фенотипических корреляций при муковисцидозе.”

Прочтите и возьмите себе на заметку, особенно если вы молодые люди
В России уже много лет проводится массовое обследование новорожденных для выявления у них нескольких наследственных заболеваний. Такое обследование проводится во многих странах и называется скринингом новорожденных или неонаталъным скринингом.
Целью скрининга новорожденных является, конечно, не само выявление новорожденных с еще не проявившимися наследственными заболеваниями, а их лечение, которое позволяет предотвратить появление клинических симптомов, во многих случаях весьма тяжелых, или даже фатальных. В результате рано начатого и аккуратно проводимого лечения вместо тяжело больных детей, а затем подростков и взрослых, получаются здоровые люди, полноценные члены общества, нередко являющиеся гордостью семьи.
Скрининг новорожденных в России ведется в отношении 5 наследственных и врожденных заболеваний: фенилкетонурии, гипотиреоза, галактоземии, адрено-гениталъного синдрома и муковисцидоза.
ЧТО ТАКОЕ МУКОВИСЦИДОЗ?
Муковисцидоз — это наследственное заболевание, обусловленное изменением (мутацией) в гене, который отвечает за синтез белка, осуществляющего в клетках функцию канала для ионов хлора. Из-за нарушения функции этого канала слизь и другие секреты становятся очень густыми и вязкими в легких, поджелудочной железе и других органах. Это приводит к развитию в легких хронической инфекции, повреждающей легочную ткань; нарушению переваривания пищи, поскольку ферменты поджелудочной железы не могут попасть в кишечник, и другим клиническим проявлениям.
КАК НАСЛЕДУЕТСЯ МУКОВИСЦИДОЗ?
Муковисцидоз наследуется по аутосомно-рецессивному типу, когда больные в семье появляются только в одном поколении. Схема такого наследования приведена на рисунке, на котором изображен фрагмент родословной семьи, в которой родился ребенок, больной муковисцидозом. На родословной мужчины обозначены квадратиком, а женщины — кружочком. Внутри этих квадратиков и кружочков нарисована только одна хромосома (из 23 пар, имеющихся у человека), несущая нормальный или мутантный ген муковисцидоза, который помечен черной точкой.
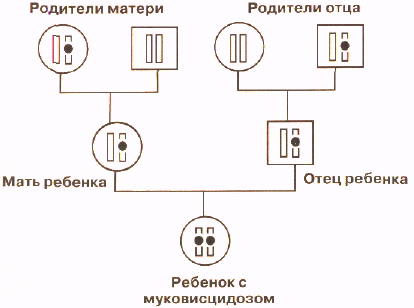
На рисунке для простоты изображена только хромосома, содержащая ген, мутации в котором вызывают муковисцидоз. У ребенка в обеих хромосомах содержится мутантный ген и поэтому он болен. У каждого из родителей мутантный ген содержится только в одной хромосоме, а вторая хромосома нормальная и поэтому они здоровы. Такие люди, которые имеют один нормальный и один дефектный ген называются носителями мутантного гена. У бабки по матери мутантный ген также имеется только в одной хромосоме, как и у деда со стороны отца. Они, как и родители ребенка, здоровы, но передали хромосомы, содержащие мутантный ген, своим детям. У вторых деда и бабки обе хромосомы содержат только нормальный ген. Таким образом, при рецессивном наследовании болен только тот член семьи, который получил от своих родителей обе хромосомы, несущие мутантный ген. Все остальные члены семьи здоровы, в том числе и те, кто является носителем мутантного гена. На схеме родословной видно, что у родителей больного ребенка могут еще появиться больные дети. Вероятность появления больного ребенка в семьях, в которых родители являются носителями мутантного гена, составляет 1/4 или 25%. Эта вероятность не меняется от числа больных или здоровых детей в семье: для каждого следующего ребенка риск, что он будет болен, составляет 25%. Вероятность рождения здорового ребенка, обе хромосомы которого содержат только нормальный ген, составляет также 25%. А 50% детей будут иметь один нормальный и один мутангный ген, как их родители. Это означает, что при каждой беременности родители-носители имеют шансы 3 из 4 (75%) родить здорового ребенка. Многие родители больных муковисцидозом детей и их родственники, первый раз встретившись с врачом-генетиком, настойчиво повторяют, что у их ребенка не наследственное заболевание, так как в их семье ни у кого из родственников никогда не было такого заболевания. Только объяснение, что правила наследования бывают разные, и не редко больной с наследственным заболеванием может быть единственным в семье, позволяют им понять с какой ситуацией они столкнулись.
КАКИЕ ПОРАЖЕНИЯ В ОРГАНИЗМЕ ВЫЗЫВАЕТ МУКОВИСЦИДОЗ?
Заболевание обычно начинается в раннем возрасте. Клиницисты различают три основные формы муковисцидоза: легочную, кишечную и смешанную. Самой частой из них является смешанная форма. Она встречается примерно у 80% больных муковисцидозом. Легочная форма муковисцидоза проявляется хроническим обструктивным бронхолегочным процессом. Из-за того, что мокрота у больных густая и вязкая, она не отхаркивается. Большое количество белка в мокроте делает ее хорошей средой для развития разных микробов, в том числе стафилококков и синегнойной палочки. Развивается хронический воспалительный процесс, приводящий к разрушению легочной ткани. Кровь больных плохо насыщается кислородом, из-за чего начинают страдать сердце, печень и другие . органы. Лечение больных с легочной формой муковисцидоза требует применения мощных антибиотиков в больших дозах. При кишечной форме муковисцидоза нарушается процесс переваривания пищи, так как ферменты поджелудочной железы, расщепляющие белки и жиры, не попадают в кишечник вследствие закупорки протоков железы. Больные отстают от своих родственников в росте и весе. Основное лечение кишечной формы заключается в приеме ферментов поджелудочной железы. Эффективность этого лечения легко контролируется по количеству жира в кале ребенка. Препараты поджелудочной железы должны даваться в таком количестве, чтобы жира в кале не было. При смешанной форме муковисцидоза кишечные проявления муковисцидоза усугубляют поражение легких. Лечение смешанной формы наиболее сложное. У больных муковисцидозом, не получающих необходимого лечения, продолжительность жизни, как правило, относительно короткая.
СУЩЕСТВУЮТ ЛИ ТЕСТЫ НА РАННЕЕ ВЫЯВЛЕНИЕ МУКОВИСЦИДОЗА?
Больные с муковисцидозом могут быть выявлены с помощью скрининга новорожденных. В отличие от других наследственных болезней, выявляемых при скрининге, при которых раннее выявление заболевания позволяет его настолько эффективно лечить, что можно вообще избежать каких-либо клинических проявлений заболеваний, скрининг новорожденных при муковисцидозе преследует несколько иную цель. Врачи считают, что если муковисцидоз выявляется у новорожденного, и его начинают сразу же лечить, то ребенок нормально развивается физически и умственно. У него в меньшей степени будут поражаться легкие и другие органы, увеличится продолжительность жизни, которая в настоящее время, благодаря адекватному лечению, составляет в развитых странах более 35 лет. Скрининг на муковисцидоз начинается с того, что у новорожденного в родильном доме перед выпиской берут из пятки несколько капель крови, которую наносят на специально для этой цели используемую фильтровальную бумагу. Кровь высушивается, и такой бланк, на котором указана фамилия новорожденного и ряд других сведений, необходимых для его идентификации, переправляется в лабораторию региональной медико-генетической консультации. В лаборатории проводят специальное исследование, которое позволяет выявить новорожденных, у которых есть подозрение на муковисцидоз. В этом случае ребенок через педиатра вызывается в лабораторию на повторноетестирование. Родителям педиатр сообщает, что первый тест на муковисцидоз у их ребенка оказался ненормальным. У них появляется повод для беспокойства. Поэтому повторное тестирование образца крови у младенца, которое является очень важным, нужно сделать как можно быстрее. В большинстве случаев при повторном исследовании тест на муковисцидоз оказывается нормальным. Это означает, что результат первого исследования был неверный (его называют ложноположительным). Причины этого могут быть разными и связанными как с состоянием младенца, так и с какой-то ошибкой лаборатории. Этот результат, свидетельствующий о том, что у ребенка нет муковисцидоза, сразу же сообщается родителям, чтобы снять с них чувство страха.
ЧТО ДЕЛАТЬ, ЕСЛИ И ПОВТОРНЫЙ ЛАБОРАТОРНЫЙ ТЕСТ НА МУКОВИСЦИДОЗ ОКАЗАЛСЯ ПОЛОЖИТЕЛЬНЫМ?
Если и второе лабораторное исследование для выявления муковисцидоза оказалось положительным, то, в отличие от других скринируемых наследственных болезней, это еще не означает, что у ребенка есть муковисцидоз, хотя вероятность такого диагноза является высокой. Семья, в которой у ребенка повторно подтвердился положительный тест на муковисцидоз, приглашается на прием к врачу-генетику в медико-генетическую консультацию. Здесь семье объясняют, что собой представляет муковисцидоз и организуют прием у клиницистов, являющихся специалистами в диагностике и лечении муковисцидоза. Теперь наблюдение за ребенком берет на себя группа клиницистов. Для подтверждения диагноза муковисцидоза младенцу проводят так называемый лотовый тест. Это совершенно безобидный и безболезненный тест можно провести младенцу, начиная с возраста 3-4 недели жизни. Если лотовый тест оказывается отрицательным, то ребенок считается здоровым, хотя за ним клиницисты еще будут наблюдать некоторое время. Если же потовый тест оказался положительным, то диагноз муковисцидоза считается установленным, даже до появления каких-либо клинических проявлений заболевания. В этом случае врачи назначат ребенку лечение, которое надо будет строго соблюдать. Ребенок будет периодически обследоваться группой специалистов для постоянного контроля за состоянием его здоровья.
МОЖНО ЛИ ПОМОЧЬ СЕМЬЕ, В КОТОРОЙ ПОЯВИЛСЯ БОЛЬНОЙ С МУКОВИСЦИДОЗОМ, ИМЕТЬ ЗДОРОВЫХ ДЕТЕЙ?
Да, и довольно успешно. Для муковисцидоза возможна дородовая диагностика. Первым шагом в этом направлении является обращение в медико-генетическую консультацию, где врач-генетик определяет показания и возможные методические подходы к дородовой диагностике в каждом конкретном случае. Сама процедура заключается в том, что во время беременности в сроке 9-11 недель или 16-18 недель врач акушер-гинеколог проводит забор очень небольшого количества клеток плода и направляет этот материал в специальную лабораторию пренатальной диагностики. В этой лаборатории врачи лаборанты-генетики проводят молекулярную диагностику, т.е. определяют наличие или отсутствие мутации в гене, отвечающем за муковисцидоз. В случае положительного результата семья решает вопрос о прерывании беременности больным плодом или настраивается на появление еще одного больного ребенка. Это право выбора остается за семьей.
Муковисцидоз – наследственное врожденное заболевание, поражающее экзокринные железы и проявляющееся выработкой избытка слизистого секрета в организме. Из-за повышенного количества слизи страдают функции многих органов, и ребёнок с муковисцидозом нуждается в пожизненной терапии. Как выявляют муковисцидоз у детей? Рассказываем про симптомы, особенности диагностики и современные методы лечения.
Причины муковисцидоза
Патология развивается при наличии специфических мутаций в гене белка, ответственном за транспорт электролитов в протоках желез внешней секреции. Искажение генетической последовательности провоцирует возникновение структурных и функциональных нарушений синтеза белка, из-за чего слизь становится густой и плохо способной проходить через протоки. Возникает застой слизи, оптимальная среда для развития воспалительных процессов.
Муковисцидоз – заболевание наследственное, но родители могут не знать о носительстве мутации. Предупредить рецессивное наследование может консультация генетика, обследование до зачатия, а также репродуктивные технологии с тестированием эмбрионов при экстракорпоральном оплодотворении.
Формы муковисцидоза: какие органы страдают?

Выделяют несколько форм муковисцидоза в зависимости от выраженности поражения определенного органа: легочная форма, кишечная и смешанная. Кроме того, современная классификация учитывает также печеночную форму муковисцидоза, мекониальную непроходимость кишечника новорожденных, атипичный муковисцидоз – когда страдает только один орган, и стертую форму, симптомы которой могут долго оставаться нераспознанными.

Факт!
В четырех из пяти случаев развивается смешанная легочно-кишечная форма.
Симптомы у детей: на что обращать внимание
Муковисцидоз у детей разного возраста проявляется различными симптомокомплексами.
Период новорожденности и младенчества
Чаще всего болезнь проявляет себя наличием сильного кашля (нередко до рвоты), одышкой, удушьем. Муковисцидоз в менее выраженной форме может проявляться длительным восстановлением массы тела, недостатком набора веса.
Еще один симптом, характерный для муковисцидоза у новорожденных, – закупорка кишечника меконием. Мекониальный илеус, непроходимость первородного кала, встречается в одном случае из пяти при раннем начале заболевания.
Важно!
Без срочного оказания помощи мекониальный илеус приводит к жизнеугрожающим состояниям: завороту кишок или прободению кишечника.
В возрасте старше 10 суток от рождения частыми признаками муковисцидоза у детей становятся атония (вялость), отказ от сосания, избыток газов, длительная желтуха новорожденных, рвота желчью. Кожа сухая, бледная, недостаточно упругая, на вкус соленая (этот признак лег в основу старейшего лабораторного теста на муковисцидоз).
Муковисцидоз у грудных детей
По информации экспертов Союза педиатров России, муковисцидоз чаще всего выявляют у детей в возрасте до двух лет. В последние годы благодаря введению скрининга на это заболевание в обязательный список анализов в родильном доме (скрининг «пяточка») возраст постановки диагноза снизился.
Факт!
Чаще всего первые симптомы у грудных детей проявляются в возрасте полугода после введения прикорма или перевода ребёнка с грудного молока на смесь.
Симптомы муковисцидоза могут быть похожи на длительные и частые респираторные или кишечные инфекции, регулярные нарушения пищеварения. У грудных детей болезнь проявляется следующими признаками:
- Появление густого кала с жирным блеском и неприятным запахом;
- Задержки физического, а затем психоэмоционального развития;
- Одышка, кашель, приступы рвоты;
- Сухая кожа с сероватым оттенком.
В раннем детстве муковисцидоз у детей может провоцировать частые бронхиты или пневмонии с переходом в хронический бронхолегочный воспалительный процесс, из-за чего грудная клетка деформируется, становится килевидной, воронкообразной, бочкообразной формы.
Кишечная форма проявляется менее ярко в первые месяцы, а порой и годы жизни: болезнь вызывает ферментную недостаточность из-за закупорки протоков поджелудочной железы, что приводит к смазанной симптоматике, под которую подпадают многие диагнозы.
Факт!
В России муковисцидоз диагностируется у одного из 9 000 новорожденных детей.
Дошкольный и старший школьный возраст: проявления муковисцидоза

В данных возрастных группах чаще всего наблюдаются следующие симптомы муковисцидоза:
- Частые приступы кашля с выводом гнойной мокроты;
- Одышка;
- Разжижение каловых масс;
- Случаи обезвоживания;
- Выпадение прямой кишки, непроходимость кишечника;
- Изменение формы фаланг пальцев на руках («барабанные палочки»);
- Хронические синуситы и т. д.
Насторожить должны также панкреатит и признаки страдания печени у ребёнка, развитие сахарного диабета.
Муковисцидоз в подростковом периоде
В период пубертата к перечисленным выше проявлениям заболевания добавляется задержка полового развития, патологическое снижение выносливости при физических нагрузках. Поражение печени может привести к циррозу, асциту, расширению вен пищевода, часты гастриты, язвы желудка, воспалительные процессы в пищеводе и учащенная дефекация.
Диагностика муковисцидоза в России
- Первый этап диагностики в последние годы проводится в родильном отделении в рамках скрининга новорожденных, обязательной медицинской процедуры. Болезнь может подтвердиться на разных этапах скрининга.
- Первый этап – забор капли крови из пяточки ребёнка на 4-7 сутки после рождения.
- Второй этап (21-28 сутки после рождения) проводят при повышенном количестве иммунореактивного трипсина в высушенном пятне крови.
- Если на втором этапе результаты скрининга опять положительные, требуется потовая проба и генетический анализ, так как скрининг на муковисцидоз может при определенных условиях давать ложноположительный результат.
Третий этап позволяет полностью подтвердить или опровергнуть диагноз. В диагностический план также могут включаться рентгенография грудной клетки, бронхоскопия, бронхография, копрограмма, анализ мокроты и т. д.
Лечение муковисцидоза: современные методы терапии

Муковисцидоз на данный момент – пожизненное неизлечимое заболевание, требующее постоянной и комплексной терапии для облегчения состояния и профилактики осложнений.
Базовая терапия включает медикаментозные препараты – антибиотики, муколитики, ферментные препараты, витамины; регулярные упражнения дыхательной гимнастики, диету, специальные аппаратные методы, облегчающие вывод слизи из легких и бронхов и так далее. Тяжелое поражение легкого или печени требует трансплантации.
Комплекс мер, направленных на вывод мокроты, назначается пожизненно и проводится ежедневно. В него входят аэрозольные ингаляции, лечебная гимнастика, вибрационный массаж, постуральный дренаж. Упражнения и постуральный дренаж показаны не менее одного раза в день, вибрационный массаж проводят от трех раз в сутки.
Из спектра муколитических препаратов отдают предпочтение дорназе альфа, гипертоническому раствору натрия хлорида, ингаляционному маннитолу и некоторым другим препаратам.
Дорназа альфа – базовый муколитический препарат в терапии муковисцидоза. Ингаляции с препаратом назначают сразу же после положительного результата диагностики.
Дорназа альфа – фермент, аналог дезоксирибонуклеазы I (ДНКазы I), в норме присутствующей в организме каждого человека в достаточном количестве. Генно-инженерный вариант ДНКазы I, дорназа альфа, применяется в симптоматической терапии заболевания почти 30 лет, и по оценке специалистов считается наиболее эффективным и безопасным средством для облегчения состояния и профилактики инфекционно-воспалительных процессов, возникающих при застое секрета.
Применение дорназы альфа вызывает разрушение внеклеточной ДНК, а значит, эффективно разжижает скопившуюся мокроту, облегчая ее вывод. Препарат достоверно снижает смертность пациентов на 15% и является незаменимой частью терапии муковисцидоза. До его разработки у врачей и пациентов не было достаточно надежного средства для эффективной муколитической терапии.
Важно!
Все больные муковисцидозом в РФ имеют право на обеспечение дорназой альфа за счет государства.
Федеральные клинические рекомендации по оказанию медицинской помощи детям с кистозным фиброзом (муковисцидозом) / Баранов А.А., Намазова-Баранова Л.С., Симонова О.И. и др. – 2020
Муковисцидоз у детей / Ивкина С.С., Кривицкая Л.В., Латохо Т.А. // Проблемы здоровья и экологии. – 2015 – №4 (46).
Эффективность и безопасность биоаналогичного лекарственного препарата Тигераза® (дорназа альфа) при длительной симптоматической терапии пациентов с муковисцидозом: результаты клинического исследования III фазы / Е. Л. Амелина, С. А. Красовский, Д. И. Абдулганиева, и др. // Пульмонология – 2019 – Том 29, № 6
№ 10 (150) декабрь, 2016
PfMfDUUM
привслжье
[ Консилиум ]
♦ ПЕДИАТРИЯ
Н.Б. МЕРЗЛОВА, В.В. ШАДРИНА, ФГБОУ ВО «Пермский государственный медицинский университет имени академика ЕА Вагнера»
Нерешенные проблемы диагностики муковисцидоза в педиатрии
Муковисцидоз — наследственное заболевание, причиной которого являются мутации в гене CFTR (Cystic fibrosis transmembrane regulator — муковисцидозный трансмембранный регулятор проводимости). В результате, у больных муковисцидозом секреты экзокринных желез имеют повышенную вязкость, что определяет патогенез заболевания. При этом страдают жизненно важные органы и системы, вызывая тяжелые осложнения, снижающие качество и продолжительность жизни.
Частота встречаемости муковис- Если ранее продолжительность жизни цидоза варьирует в различных больных муковисцидозом была не более 5
регионах России, в среднем составляет 1:10000 новорожденных. В различных странах частота встречаемости муковисцидоза от 1:1800 в Ирландии до 1:100000-350000 новорожденных в Японии. В Пермском крае частота встречаемости муковисцидоза, по данным скрининга, 1:12713 новорожденных.
В России с 2011 года ведется регистр пациентов муковисцидозом. Регистр был создан для обобщения клинико-эпидемиологической ситуации муко-висцидоза в стране и оценки качества оказания медицинской помощи пациентам. По данным Федерального регистра, в 2014 году насчитывался 2131 пациент с муковисцидозом из 74 регионов-субъектов Российской Федерации. Из всех зарегистрированных пациентов России в Москве наблюдались 334 больных и Московской области — 171 пациент, в Пермском крае — 71 больной. Так, большинство пациентов муковисцидозом наблюдались в региональных центрах.
лет, то в настоящее время уже растет количество взрослых пациентов. В Пермском крае в 2015 году наблюдалось 74 пациента, из них 41 в возрасте до 18 лет и 33 -старше 18 лет.
Увеличение продолжительности жизни пациентов в последние годы достигнуто благодаря ранней диагностике заболевания и отработанной тактике наблюдения детей, выявленных при скрининге на муковисцидоз.
Продолжительность и качество жизни больных муковисцидозом увеличилось и благодаря современным методам диагностики, лечения, диспансерного наблюдения и профилактике осложнений заболевания (рис. 1). В настоящее время больные наблюдаются в региональных специализированных центрах, где осуществляется комплексное обследование и своевременная коррекция лечения.
В некоторых странах скрининг новорожденных на муковисцидоз проводится с 70-х годов, в России — с 2006 года внедрен неонатальный скрининг на муковис-цидоз — исследование крови на иммуно-
проф. Н.Б. Мерзлова
реактивный трипсиноген (ИРТ), благодаря чему стала возможной диагностика муковисцидоза с периода новорожденно-сти, что позволило своевременно начать лечение пациентов. В Пермском крае с внедрением скрининга также отмечена преимущественно ранняя диагностика заболевания (рис. 2).
Так, в Пермском крае при проведении скрининга за 10 лет выявлено 28 детей, диагноз муковисцидоз им был установлен в возрасте до 2 месяцев жизни (таблица 1).
Метод определения иммуно-реак-тивного трипсиногена имеет высокую чувствительность и специфичность. По данным Регистра, в 2014 году впервые было выявлено 123 пациента. Из них благодаря проведению скрининга выявлено 92 больных (74,8% всех случаев). Однако существу-
H PEMtOUUM
_привслжье
№ 10 (150) декабрь, 2016
КОНСИЛИУМ. ПЕДИАТРИЯ
ют данные о ложноотрицательных случаях. По данным Регистра 2014 года, отмечены случаи ложноотрицательных результатов скрининга новорожденных на муковисци-доз в Московской, Ярославской, Тульской, Самарской, Омской, Нижегородской, Ленинградской и Брянской областях, в Алтайском крае, республиках Чувашии и Татарстан. В 2016 году в Пермском крае
выявлена больная муковисцидозом, имевшая нормальные показатели иммуно-реактивного трипсиногена в периоде ново-рожденности, с одной «мягкой» мутацией в генотипе. Ложноотрицательные результаты ИРТ как правило, обусловлено наличием мягких мутаций, без выраженного поражения поджелудочной железы, вследствие чего нет повышения ИРТ в крови. Это сни-
жает чувствительность метода для «мягких» генотипов.
Потовый тест остается «золотым стандартом»» диагностики муковисцидоза. Показатели теста отражают функцию хлорных каналов. В настоящее время в большинстве центров в России потовый тест проводится методом проводимости на аппарате «Нанодакт». Благодаря внедрению аппаратного метода потового теста появилась возможность обследовать новорожденных и маловесных детей. Однако, некоторые дети и взрослые имеют сомнительные результаты потовых тестов, они входят в группу риска по муковисцидозу и могут иметь «мягкие генотипы» или являться носителями одной мутации в гене CFTR.
Ген CFTR в настоящее время достаточно хорошо изучен. В гене CFTR выявлено более 1900 мутаций. В связи с появлением лекарственных препаратов, способных влиять непосредственно на проводимость хлорных каналов, роль генетических методов определения мутаций в гене CFTR резко возросла. В последние годы активно внедряется методика скрининга ИРТ/ДНК, причем, с активным поиском мутаций в гене CFTR, благодаря чему удается обнаружить множество ранее неизвестных мутаций в гене CFTR (Hughes E.E., Stevens C..F, Saavedra-Matiz CA et all., 2016).
Предложены различные панели для диагностики муковисцидоза в России, которые включают до 50 мутаций, наиболее часто встречающихся мутаций в популяции. Самой распространенной мутацией в гене CFTR во всех регионах остается F508 del, кроме Республики Чувашия, где самой частой мутацией явилась Е92К. Исследование практически в каждом регионе среди генетически обследованных пациентов остаются больные с неиденти-фицированными мутациями — до 46% аллелей в Удмуртской республике и 49% аллелей в Пермском крае. Регионы граничат между собой. В обоих регионах проживают представители более 100 различных национальностей, что еще больше затрудняет ДНК-диагностику заболевания. Существуют региональные популяции, имеющие свои генетические особенности, что затрудняет обнаружение мутаций в гене CFTR при проведении обследования на стандартных панелях. Кроме того, в России исследова-
[КОНСИЛИУМ. ПЕДИАТРИЯ]
№ 10 (150) декабрь, 2016
ние мутаций в гене CFTR проводится лишь в некоторых лабораториях. Существуют технические сложности с транспортировкой образцов крови из некоторых региональных центров в лаборатории ДНК-диагностики.
По данным регистра больных муковисцидозом в Российской Федерации 2014 года, наблюдаются больные муковисцидозом, имеющие 121 различную мутацию. У больных Пермского края были определены различные мутации в гене CFTR (табл. 2).
В настоящее время внедрен метод секвенирования всей кодирующей области гена CFTR, при помощи которого появилась возможность выявлять до 98% мутаций в гене CFTR. Однако метод имеет высокую стоимость и недоступен для большинства семей, где имеются больные муковисцидозом.
Кроме того, даже при обнаружении редких мутаций возможны сложности с их интерпретацией в связи с отсутствием в базах CFTR1 и CFTR2.
В сложных диагностических случаях в некоторых странах проводится определение разности назальных или ректальных потенциалов, позволяющих определять функцию хлорных каналов.
Внедрены современные комплексные методы лечения. Микрокапсулированные ферменты поджелудочной железы позволяют значительно улучшить нутритивный статус больных. В лечении больных применяется дорназа альфа, безопасность которой доказана и у больных раннего возраста. Внутривенные и ингаляционные антибактериальные препараты способствуют предупреждению и своевременному купированию обострения инфекционного процесса в легких. В тяжелых случаях применяется трансплантация легких и печени.
Таким образом, несмотря на значительные успехи в диагностике заболевания, выявление мягких генотипов муковисцидоза затруднено, что требует дальнейшего изучения и совершенствования методов лабораторной диагностики заболевания.
р п fMEOLIUM ривслжье
Сиалор
Антибактериальные свойства Д Удобная форма выпуска Срок хранения 2 года*
т&мышлении еиде
www.siaior.ru
ргкгммл. Гигмничеода срсдс reo. Не является лечре гв<?м
Bionorica
RENEWAL
ТАБЛИЦА 1 Частота встречаемости MB в Пермском крае по данным скрининга на MB
Год наблюдения Количество детей, родившихся живыми Количество больных МВ, родившихся в данном году Частота МВ по данным скрининга
2006 30266 1 1:30266
2007 32898 1 1:32898
2008 35519 4 1:8880
2009 36139 2 1:18069
2010 37432 2 1:18716
2011 36096 4 1:9024
2012 32597 3 1:10865
2013 38110 4 1:9527
2014 38954 3 1:12985
2015 37969 4 1:9492
Всего 355980 28 1:12713
ТАБЛИЦА 2 Аллельная частота мутаций в гене СЩ, обнаруженных у пациентов МВ в Пермском крае, п=73
мутация Частота, %
[deltalF508 33,3
CFTRdele2, 3 4,8
N1303K 3,5
2143delT 3,5
3272- 16T- >A 2,8
L138ins 2,1
3849+ 10kbC- >T 0,7
3849G->A 0,7
2789+ 5G>A 0,7
621+ 1G- >T 0,7
3944delGT 0,7
S1196X 0,7
E92K 0,7
c.580G>A 0,7
Unknown 44,4
Всего аллелей 100
0 Ускоряет выведение трудноотделяемой мокроты 0 Оказывает противовоспалительное действие 0 Обладает противовирусной активностью
§
www.bionorica.ru ^
‘ Бронхипрет* сироп — для взрослых и детей от 3-х месяцев; Бронхипрет* ТП таблетки — для взрослых и детей старше 12 лет.
431 просмотр
5 ноября 2022
Здравствуйте, ребёнку 3 недели, сегодня позвонили из поликлиники,повышение итр(подозрение на муковисцидоз) в 2 раза!!!норма 65,у нас 130!!!я очень переживаю, такое повышение говорит о диагнозе???сегодня взяли повторный анализ крови,пока будет результат, я вся изведусь…Ребёнок на гв,стул частый,после каждого кормления,желто зелёный без запаха сильного,не срыгивает,начиталась, теперь и лоб кажется солоноватый у ребенка…набрал за 3 нед 500 грамм.
На сервисе СпросиВрача доступна бесплатная консультация педиатра онлайн по любой волнующей Вас проблеме. Врачи-эксперты оказывают консультации круглосуточно. Задайте свой вопрос и получите ответ сразу же!

Педиатр
Здравствуйте, нужно набраться терпения , перепроверить ещё раз. Прибавка впринципе нормальная для 3х недель. Стул обильный?
Наталья, 5 ноября 2022
Клиент
Ольга, он после каждого кормления, в среднем около наверное чайной ложки,но у меня и со страшим ребёнком в первые месяцы на гв такой частый стул бвл,поэтому как то не запозртла ничего. СКАЖИТЕ пожалуйста на вашей практике такое повышение может дать ложноположит.результат и часто ли они?
Педиатр
На гв это норма. Вообще при муковисцидозе стул обильный и ребёнок плохо прибавляет в весе. На моей практике был такой случай, когда не подтвердили МВ да.

Педиатр, Дерматолог, Венеролог, Детский
Здравствуйте, Наталья
В данном случае не исключена ошибка, но в любом случае нужно пересдать и ждать результата. Достаточно часто бывает дефект сбора, поэтому раньше времени не переживайте
Наталья, 5 ноября 2022
Клиент
Дарья Николаевна, СКАЖИТЕ пожалуйста на вашей практике такое повышение может дать ложноположит.результат и часто ли они?
Педиатр, Дерматолог, Венеролог, Детский
Да, на моей практике было.
Не очень часто, пару раз точно было

Педиатр
Здравствуйте, нужно дождаться повторного результата, терпения🙏🏻Стул такой частоты, консистенции и цвета в норме у всех младенцев
Наталья, 5 ноября 2022
Клиент
Алина Александровна, СКАЖИТЕ пожалуйста на вашей практике такое повышение может дать ложноположит.результат и часто ли они?
Педиатр
Да, может, бывают ошибки лаборатории. Про частоту таких случаев однозначно нельзя сказать, но раньше времени не переживайте, малыш чувствует беспокойство мамы и тоже может беспокоиться

Педиатр
Здравствуйте!
Делать выводы пока ещё рано. Поэтому и сделали повторный забор крови, чтобы исключить неправильный забор и лабораторные ошибки.
Самое главное сейчас успокоиться и ждать результата. Будем надеяться, что это ошибка.
Стул пенится?
Наталья, 5 ноября 2022
Клиент
Эндже, СКАЖИТЕ пожалуйста на вашей практике такое повышение может дать ложноположит.результат и часто ли они?
Стул не пенится
Педиатр
Положительный результат скрининга говорит о том, что есть подозрение на муковисцидоз.
Это ещё не означает 100% наличие. Часто бывают каждых ложноположительные результаты, 3-6 случаев положительного результата только один оказывается действительно больным муковисцидозом, а оставшиеся — это ложноположительные результаты.
В моей практике ещё не было положительных результатов.
Обычно, для постановки диагноза решающее значение имеет потовый тест,

Педиатр
Здравствуйте) нужно дождаться повторного результата, в лабораториях бывают ошибки.

Педиатр
Здравствуйте Наталья, судя по описанию абсолютно здоровый малыш, набор в весе хороший , надеюсь результат ошибочный, наберитесь терпения не переживайте, будем ждать повторный результат, как придут отпишитесь. Будьте здоровы!
Оцените, насколько были полезны ответы врачей
Проголосовало 0 человек,
средняя оценка 0
Что делать, если я не нашел ответ на свой вопрос?
Если у Вас похожий или аналогичный вопрос, но Вы не нашли на него ответ — получите свою онлайн консультацию врача.
Если Вы хотите получить более подробную консультацию врача и решить проблему быстро и индивидуально — задайте платный вопрос в приватном личном сообщении. Будьте здоровы!
МУКОВИСЦИДОЗ: Проблемы и пути их решения.
 Пути решения проблем, возникающих у пациентов с муковисцидозом
Пути решения проблем, возникающих у пациентов с муковисцидозом
Муковисцидоз (МВ) — моногенное, аутосомно-рецессивное генетическое заболевание, характеризующееся поражением экзокринных желез жизненно важных органов и имеющее обычно тяжелое течение и прогноз. Муковисцидоз является важной медико-социальной проблемой. Раньше это была проблема педиатров, в настоящее время все большее число специалистов разного профиля вовлекается в ее решение. Если в 50-годах более 60% больных МВ умирало в возрасте до 1 года, то в настоящее время более 50% наблюдаемых в США больных МВ старше 18 лет[1]. По данным J.Dodge [2] прогностичекая выживаемость больных, рожденных по неонатальному скринингу 50 лет и старше.
С 2011 года специалистами Российского детского (научно-клинического отдела муковисцидоза ФГБНУ Медико-генетического научного центра) и взрослого (лаборатория муковисцидоза ФГБНУ НИИ пульмонологии ФМБА России) центров муковисцидоза был создан и интегрирован в Европейский, регистр пациентов с муковисцидозом РФ. По данным анализа регистра за 2014 год, медина выживаемости московских пациентов составила 39,7 лет, что соответствует ведущим европейским и американским центрам. К сожалению, общие данные по выживаемости российских пациентов в стране низкие, что обусловлено рядом имеющихся проблем.
Распространенность МВ варьирует в зависимости от популяции. В большинстве стран Европы и Северной Америки МВ она колеблется от 1:2000 до 1:5000 новорожденных. Расчетные данные, полученные в Медико-генетическом научном центре и в Минздравсоцразвития РФ (неонатальный скрининг), свидетельствуют о более низкой частоте МВ в России — 1:8000-12000 новорожденных. Большую роль в улучшении помощи больным МВ внесли как организация в нашей стране специализированных Российского детского (дата организации — 1990г) , взрослого (организован в 1992г) и региональных центров по диагностике (в том числе пренатальной), лечению и реабилитации больных муковисцидозом, включение муковисцидоза с 2006-2007 г в программу неонатального скрининга (приказ Минздравсоцразвития России от 22 марта 2006 г. N 185 «О массовом обследовании новорожденных детей на наследственные заболевания»), так же действующие Приказы Минздравсоцразвития по ряду льгот для инвалидов детства, больных МВ. Разработаны стандарты лечения и наблюдения пациентов с МВ. Обеспечение больных МВ лекарственными препаратами осуществляется по программе «7 нозологий» по Распоряжению Правительства РФ (от 2 октября 2007г. № 1328-р), которая, в настоящее время предусматривает только один препарат – дорназа альфа и из средств региональных бюджетов (ферменты, ингаляционные препараты, антибактериальные лекарственные средства и др)[3].
На активном диспансерном наблюдении в Российском детском и взрослом центрах, выполняющих роль межрегиональных, находится около 900 больных МВ, из них 44% старше 18 лет.
В центре апробируются и успешно применяются новейшие медицинские препараты, современные лечебно-реабилитационные режимы и технологии, которые позволяют подавляющему большинству больных вести полноценный образ жизни. Однако существующие проблемы зачастую препятствуют эффективной диагностике, наблюдению и лечению таких пациентов. Все проблемы делятся на три большие группы – проблемы диагностики, лекарственного обеспечения и организации команды врачей и условий для оказания профессиональной медицинской помощи.
Проблемы в диагностике муковисцидоза
Неонатальный скрининг.
С 2006 г. в ряде регионов, а с первого января 2007 г. во всех субъектах Российской Федерации (РФ) МВ был включен в перечень наследственных заболеваний, подлежащих обязательному неонатальному скринингу в рамках национального приоритетного проекта «Здоровье». В настоящее время в нашей стране скрининг проводится в 4 этапа (табл.1).
Таблица 1. Этапы неонатального скрининга в РФ
| I ЭТАП | На 3-4 день у доношенного (7-8 – у недоношенного) определение ИРТ 1 в высушенной капле крови |
| II ЭТАП | При положительном результате (более 70 нг/мл) на 21-28 день — повторный тест — ИРТ 2 |
| III ЭТАП | При положительном результате ИРТ 2 (более 40 нг/мл) — потовая проба |
| IV ЭТАП | При пограничном результате потовой пробы — ДНК-диагностика |
I. Проблемы при диагностике МВ по неонатальному скринигну :
1. Отказ родителей от проведения дообследования ребенка, при положительных данных неонатального скрининга
В Москве за период с 2006 г. по октябрь 2010 г. по программе неонатального скрининга было обследовано 552 342 новорожденных. Из них гипертрипсиногенемия после 2 этапа скрининга (повышение ИРТ 2) сохранялась у 905 (16,9%). Все семьи, согласно протоколу скрининга, были приглашены в московский центр МВ для дальнейшего обследования. Однако, на проведение потовой пробы явилось только 645 (71%) семей. В 29% случаев родители по тем или иным причинам отказались от дальнейшего обследования.
2. Не соблюдение сроков проведения неонатального скринига и техники.
За период с 2006 г по 2010г в Москве было выявлено по программе скрининга 54 пациента и 4 пациента с «ложноотрицательными» данными ИРТ. Вывод: имеют место нарушение техники проведения скрининга и его сроков. Вероятно, недостаточная информированность медицинского персонала поликлиник могут влиять на качество проводимой диагностики муковисцидоза.
3. Отсутствие расходных материалов для скрининга и потового теста
Специалисты из региональных центров неонатального скрининга, на мероприятиях организуемых Российским центром МВ, отмечают нерегулярное обеспечение дорогостоящими расходными материалами для проведения скрининга и потовых проб.
4 . Не верная интерпретация данных скрининга, в связи с результатами ДНК диагностики.
Одна из наиболее частых причин гиподиагностики. Например, скрининг у пациента положительный, потовая проба положительная или пограничные значения, мутаций при ДНК диагностике гена CFTR не обнаружено или обнаружена только одна, диагноз не подтверждают, что в корне не верно. Пациенты с гипертрипсиногенемией не должны быть сняты с наблюдения в профильном центре до 1 года. Положительные данные скрининга (даже при отрицательном потовом тесте и отсутствии наиболее частых мутаций при ДНК-диагностике) являются показанием для диспансерного наблюдения в течение года жизни, повторной консультации и потовой пробы в возрасте 1 года. В настоящее время, благодаря совершенствованию генетической диагностики и расширению знаний о муковисцидозе, выявлены мутации, частично сохраняющие работу хлорного канала и соответственно приводящие к отрицательным результатам потового теста, как например 3849+10kbC>T
Не обнаружение мутаций при стандартной ДНК-диагностике не дает право исключить у пациента муковисцидоз. В настоящий момент известно более 2000 мутаций в гене CFTR. Стандартная панель ДНК диагностики для поиска мутаций гена CFTR в различных региональных центрах охватывает от 8 до 35 наиболее частых мутаций, составляющих около 75% встречающихся в данной популяции, что не позволяет ориентироваться на данный метод, как на основной в постановке диагноза. В то же время, обнаружение 2 мутантных аллелей достоверно подтверждает муковисицидоз, даже при отсутствии или отрицательных данных скрининга и потового теста. В сомнительных случаях в диагностике муковисицидоза используют полногеномное секвенирование гена CFTR.
Проведение ДНК-диагностики за счет средств семьи пациента
ДНК-диагностика является очень важной в анализе возможных рисков и осложнений у пациента, применении у больных с определенными кассами мутаций разработанной в настоящее время патогенетической терапии заболевания. Вместе с тем, ДНК-диагностика является дорогостоящим обследованием и не каждая семья или пациент может оплатить его стоимость. Проведение дорогостоящего секвенирования гена при отсутствии мутаций у пациента с сомнительной клинической картиной заболевания, позволит правильно поставить диагноз или снять его при отрицательных данных полногеномного генетического секвенирования. Важность ДНК диагностики также определяется необходимостью проведения пренатальной диагностики в семье, где есть уже ребенок с муковисицидозом у родителей-носителей дефектного гена для определения наличия мутаций у плода и принятия решения о вынашивании беременности родителями.
II. Нарушение техники проведения потовой пробы
Ложные результаты потового теста выполненного как классическим методом по Гиббсону-Куку, так и анализе проводимости на аппаратах Нанодакт, Макродакт возможны при нарушении техники проведения обследования и недостаточном опыте лаборатории. Перед исследованием необходимо тщательно очистить и обезжирить кожу пациента в месте забора потовой жидкости. Помнить, что при ряде состояний потовый тест может давать ложноположительные или ложноотрицательные значения, поэтому приоритетно проводить данные исследования в профильных центрах, а при сомнительных данных отправлять на консультацию к специалистам Российского центра муковисицидоза, имеющих многолетний опыт в диагностике заболевания.
III. Диагностика групп риска
В связи с расширением знаний о муковисицидозе, специалисты центров муковисцидоза стали выявлять пациентов с мягким или моносимптомным течением заболевания, а также диагносцировать заболевание у взрослых больных. Поскольку проявления муковисицидоза многогранны, такие пациенты могут длительное время наблюдаться врачами различных специальностей с другими диагнозами. Крайне важна информированность специалистов «на местах» о группах риска для диагностики на муковисицидоз (синдром мальабсорбции, полипоз, мекониальный илеус, бронхоэктазы, сахарный диабет у пациентов с бронхолегочными заболеваниями, цирроз печени, и тд).
Пути решения проблем с диагностикой муковисцидоза.
1. Информирование о заболевании врачей и специалистов среднего звена поликлиник, пульмонологов, гастроэнтерологов, репродуктологов, гепатологов, трансплантологов, оториноларингологов о возможных группах риска и алгоритме диагностики заболевания.
2. Обучение врачей центров МВ или оказывающих помощь больным МВ на специализированных циклах (например, цикл ТУ, проводимый сотрудниками ФГБНУ «МГНЦ» и ФГБОУ «Сиб ГМУ»).
3. Введение в обязательную программу аккредитации врачей специализированных центров неонатальной диагностики сертификации по неонатальному скринингу и диагностике муковисицидоза на базе ФГБНУ «МГНЦ».
4. Необходимость раннего выявления МВ возможно при соблюдении протоколов неонатального скрининга, техники проведения потовых тестов, своевременной диагностики пациентов из групп риска.
5. Необходимо финансирование из федерального бюджета закупки оборудования и расходных материалов для проведения потовых тестов детям с гипертрипсиногенемией, выявленным по программе неонатального скрининга, а так же организация своевременной закупки расходных материалов для неонатального скрининга.
6. Информирование населения о необходимости проведения неонатального скрининга новорожденным детям и при положительных результатах, важности своевременного дообследования в условиях специализированных центров.
7. Специалисты, занимающиеся диагностикой МВ должны учитывать, что не выявление мутаций при стандартной ДНК-диагностике не может являться основанием для исключения заболевания у пациента.
8. Проведение ДНК-диагностики у выявленных по скринингу или клинической картине пациентов за счет средств регионального бюджета, та как своевременная ДНК диагностика позволит более эффективно проводить патогенетическое лечение пациентов, информирование родителей и родственников пациента о риске повторного рождения ребенка с МВ позволяет профилактировать заболевание.
Проблемы перекрестного инфицирования пациентов
За последние 20 лет теория перекрестного инфицирования среди больных МВ была доказана [4] Наиболее распространенными возбудителями заболеваний дыхательного тракта при МВ являются: Staphilococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa. Все большее значение, в последнее время, приобретают Burkholderia cepacia, Stenotrophomonas maltophilia, Achromobacter xylosoxidans, Aspergillus spp., нетуберкулезные микобактерии, грибы, MRSA. Ряд патогенов, не представляющих опасности для здоровых людей, для пациентов с МВ могут быть фатальными и способствовать быстрому регрессу функции легких. Так, например, мерам строгой контактной изоляции подлежат пациенты, имеющие в мокроте Burkholderia cepacia, MRSA, полирезистентные штаммы Pseudomonas aeruginosa, нетуберкулезные микобактерии. Из-за экономической составляющей и существующих тарифов ОМС, распространенные в мировой практике стандарты амбулаторного наблюдения и домашнего стационара для пациентов с МВ в России не выполняются, хотя являются более экономически целесообразными. Пациенты, выявленные по неонатальному скринингу зачастую госпитализируются в стационар для дообследования и назначения лечения. Такая практика подвергает высокому риску перекрестного инфицирования новорожденного серьезной патогенной микрофлорой, к которой пациенты предрасположены. Пациенты госпитализируются для планового обследования, проведения ингаляционной антибактериальной терапии. Причем, только единичные стационары имеют одноместные палаты с необходимым санузлом и раковиной для больных с МВ. Существует распространенная ошибка, что пациенты с однородной инфекцией, например синегнойной, могут госпитализироваться в одну палату. В то время, как фенотипы Pseudomonas aeruginosa могут быть разными, и пациенты заражают друг друга.
Более 20-ти лет существования Российского центра МВ (московского отделения) происходило переориентирование его работы на амбулаторное наблюдение, обследование и лечение пациентов [5]. Все это привело к значительному изменению состояния больных, минимализации рисков инфицирования пациентов фатальной микрофлорой и соответственно к значительному увеличению медианы и качества жизни таких больных [6]. Тем не менее, правила жесткого контроля за эпидемиологической обстановкой в центрах муковисицидоза, отказа от необоснованных госпитализаций и переориентирования на амбулаторное наблюдение не исключают строгого соблюдения правил по профилактике перекресного инфицирования в условиях поликлиники, а также информирования родителей о возможных рисках вне медицинских учреждений. Работа зарубежных центров МВ ориентирована на амбулаторную помощь и организацию профилактики перекрестного инфицирования, в том числе по времени посещения пациентов с различной инфекцией и четкой организации обработки помещения, оборудования, рук персонала и пациентов, использования масок.
Адекватный контроль за микробной флорой и профилактика перекрестного инфицирования не возможны без регулярного микробиологического мониторинга пациентов, который проводится 1 раз в 1 —3 месяца . К сожалению, возникает ряд проблем в идентификации микроорганизмов и организации работы лабораторий:
- Микроорганизмы (S.aureus, P.aeruginosa и др.) при хронической персистенции могут менять свой фенотип. Их становится сложно распознать в лабораториях, не специализирующихся на микробиологических анализах при МВ.
- Местные условия среды могут приводить к образованию гипермутабельных штаммов с высокой вариабельностью генотипических и фенотипических черт, включая резистентность к а/б препаратам. У одного пациента могут высеваться до 4-5 микроорганизмов.
- Отсутствие необходимых для идентификации специфических микроорганизмов дорогостоящих питательных сред и реактивов для широкого спектра антибиотиков при определении чувствительности, а так же короткое время посева (например, 3 дня) в региональных лабораториях, делает данную диагностику бесполезной.
- Ранняя диагностика инфекции бронхиального дерева затрудняется трудностью забора мокроты у новорожденных и детей раннего возраста.
Пути решения проблем, связанных с перекрестным инфицированием пациентов.
1. Госпитализации в стационар пациентов с муковисцидозом должна осуществляться только по жизненным показаниям.
2. Внедрение в практику региональных центров активного амбулаторного наблюдения и практики домашнего стационара.
3. Внесение поправки в Федеральный закон Российской Федерации от 21 ноября 2011 г. N 323-ФЗ статью 34 п.2 «Специализированная медицинская помощь оказывается в стационарных условиях и в условиях дневного стационара» специализированной помощи в амбулаторных условиях.
4. Перераспределение объемов медицинской помощи с ресурсоемкой и дорогостоящей стационарной на догоспитальные стационарозамещающие технологии, оптимизация медицинской помощи в домашних условиях, которые являются более экономически целесообразными и позволяют значительно снизить возможности перекрестного инфицирования данной категории больных.
5. Строгий микробиологический мониторинг, с учетом особенностей диагностики инфекций у больных муковисцидозом
6. Обучение специалистов и оснащение профильных микробиологических лабораторий, поскольку не идентифицированные патогены могут представлять опасность не только для пацеинтов с муковисицидозом, но и для иммуносупрессивных больных, а также для пациентов с тяжелой патологией.
7. Строгое соблюдение во всех специализированных центрах и стационарах правил по профилактике перекресного инфицирования пациентов
8. Разделение амбулаторного приема пациентов по высеваемой флоре (начало недели — S.aureus, H.influenzae, с условно-патогенной микрофлорой, конец недели — P.aeruginosa, пациентов с B.cepacia, MRSA, НТМБ принимать в отдельных помещениях и принимать меры по контактной изоляции при госпитализации в стационар).
9. Использовать в специализированных центрах одноразовые маски для пациентов, бесконтактные диспенсеры с антибактериальным средством перед кабинетами для приема, одноразовые халаты и пеленки для осмотра.
10. Обрабатывать руки, пульсоксиметр, фонендоскоп, другое оборудование антибактериальным раствором после осмотра пациента, проводить влажную дезинфекцию поверхностей и проветривать кабинет после каждого пациента.
11. Использовать современные системы обеззараживания воздуха в помещении
12. При исследовании функции внешнего дыхания обязательным является использование одноразовых антибактриальных фильтров для спирометра.
13. Госпитализировать пациентов в индивидуальные боксы, больным с МВ не разрешается посещать игровые комнаты, туалеты, ванную и столовую в отделении.
14. Ингаляционное оборудование и дыхательные тренажеры могут быть только индивидуального пользования.
Проблемы лекарственного обеспечения
1. Необходимо изменить тактику обеспечения лекарственными средствами только после госпитализации в стационар или оформления инвалидности на практику раннего назначения стандартной терапии заболевания
В ряде регионов лекарственное обеспечение новорожденных пациентов, выявленных по программе скрининга, начинают только после госпитализации в стационар или оформления инвалидности. Если состояние ребенка не является жизнеугрожающим (мекониальный илеус, дыхательная недостаточность, синдром псевдо-Барттера или др), необходимое обследование рекомендуется проводить в амбулаторных условиях. Лекарственное обеспечение, особенно ферментную терапию, необходимо назначать как можно скорее, с учетом данных копрологии и клинической картины, степени выраженности панкреатической недостаточности, поскольку нутритивный статус пациентов напрямую коррелирует с проводимой заместительной терапией панкреатическими ферментами и определяет состояние бронхолегочной системы. А не своевременное назначение ингаляционной муколитической терапии при респираторных проявлениях, может приводить к ранней манифестации поражения дыхательной системы.
2. Отсутствие своевременного и адекватного лечения жизнеугрожающих инфекций
При первичном инфицировании пациента такими фатальными микробами, как P.aeruginosa, B.cepacia, MRSA, НТМБ и проведении своевременной высокоэффективной терапии возможно добиться эффективной эрадикации патогена. В то же время, при длительном отсутствии жизненно важного лекарственного препарата( более 2 недель) риск хронической колонизации и необходимости постоянного применения дорогостоящих лекарственных средств возрастает в разы. Например, лечение пациента с хронической колонизацией золотистого стафилококка ограничивается, как правило, пероральным применением антибактериальных препаратов во время обострения заболевания и по приблизительным рассчетам затраты на антибиотики составляют около 5 тыс рублей в год. Больному с хронической колонизацией синегнойной палочки требуется постоянная ингаляционная противосинегнойная терапия и ежеквартальные профилактические курсы внутривенной антибактериальной терапии 2-3 противосинегнойными препаратами, что при рассчете в год составляет около 1,5 млн рублей на пациента. Препараты для эрадикации первичного инфицирования синегнойной палочкой (ингалиционные антибиотики – тобрамицин и колиместат натрия) применяются в течение 1 – 3 месяцев. Так, затраты на терапи. Брамитобом (Chiesi, Италия) составляют не более 100 тыс рублей. Таким образом, своевременная терапия первого высева синегнойной палочки экономически более выгодна. Для лечения B.cepacia может применяться неприрывная антибактериальная терапия 3-4 дорогостоящими антибиотиками, что увеличивает стоимость лечения в разы, даже по сравнению с пациентами с хронической синегнойной инфекцией. Замена антибактериальных препаратов дженериками, которые по стоимости от оригинальных отличаются незначительно, но,к сожалению, часто не эффективны и дают большое количество побочных эффектов.
Замена жизненно важных препаратов на дженерики, не проходившие клинические исследования и на препараты, доказавшие свою неэффективность у пациентов с муковисцидозом
Пациенты с МВ получают лекарственную политерапию в течение всей жизни, что обуславливает необходимость назначения препаратов только с доказанной клинической эффективностью и безопасностью. К сожалению, часто биодоступность и эффективность закупаемых дженериков не соответствует должной. Например, препарат для пожизненой заместительной терапии панкреатичесой недостаточности Микразим, представляющий собой микрогранулированный панкреатин, был исследован еще в 2007-2008гг в Российском центре муковисицидоза (Москва) совместно с 6 региональными центрами (Саранск, Оренбург, Казань, Саратов, Волгоград, Астрахань)[7]. Полученные результаты показали, что препарат Микразим обладает высокой частотой (26%) серьезных побочных явлений со стороны желудочно-кишечного тракта, потребовавших его отмены, и отсутствием эффективности еще у 14% пациентов. Учитывая выше изложенные данные препарат не может быть рекомендован пациентам с муковисицидозом. Тем не менее, в рамках программы бесплатного лекарственного обеспечения, препарат централизовано закупается в регинах РФ, в том числе и для московских больных. Ежемесячно врачи центра регистрируют нежелательные побочные явления (НПЯ) при использовании Микрозима.
Пример эффективного и безопасного дженерика представляет ингаляционный раствор тобрамицина — Брамитоб (Италия), являющийся копией препарата Тоби (Швейцария). Раствор Брамитоба участвовал в многочисленных международных мультицентровых плацебо-контролируемых клинических исследованиях у данного контингента больных, подтвердивших его эффективность и безопасность [8]. Эти исследования позволяют рекомендовать данный дженерик пациентам с муковисицидозом, в отличие от часто закупаемого раствора тобрамицина для ингаляций «Тобрамицин-Гобби». В настоящий момент говорить о клинической эффективности и безопасности препарата не представляется возможным. Упоминания о его безопасности и эффективности у больных муковисцидозом нет ни в одном научном зарубежном источнике. Препарат «Тобрамицин – Гобби» не получал одобрение регуляторных органов США и Европейского Союза и не прошел необходимых клинических исследований по оценке его безопасности и эффективности при МВ. Имеются данные об отсутствии эффекта данного препарата и НПЯ у пациентов с муковисцидозом в ряде регионов России.
Пути решения проблем, связанных с лекарственным обеспечением
1. Информирование профильных специалистов о необходимости назначения лекарственной терапии в максимально короткие после постановки диагноза сроки.
2. Назначение терапии согласно протоколам и стандартам ведения больных муковисцидозом
3. Исключить госпитализацию пациентов для проведения курсов ингаляционной антибактериальной терапии.
4. Своевременное обеспечение эффективными антибактериальными препаратами , доказавшими свою терапевтическую активность и безопасность у пациентов с муковисцидозом с первичным инфицированием P.aeruginosa, B.cepacia, MRSA, НТМБ
5. Возможность назначения препаратов по торговому наименованию пациентам с муковисицидозом, с учетом отсутствия у большинства дженериков данных о доказанной терапевтической эквивалентности и биодоступности (ФЗ от 12.04.2010 N 61-ФЗ (ред. от 29.12.2015) «Об обращении лекарственных средств»12.3)
6. При наличии медицинских показаний (индивидуальная непереносимость, по жизненным показаниям) по решению врачебной комиссии медицинской организации осуществляется назначение и выписывание лекарственных препаратов по торговым наименованиям (пункт 3 приложения N1 к приказу Минздрава России от 20 декабря 2012 г. N 1175н). Упростить процедуру принятия решений, которая составляет от 3-6 месяцев.
7. Соблюдение права пациента на получение качественных, безопасных и доступных лекарственных препаратов согласно статье 18 п.2 ФЗ от 21.11.2011 N 323-ФЗ (ред. от 29.12.2015) «Об основах охраны здоровья граждан в Российской Федерации».
Проблемы со подготовкой специалистов для оказания помощи больным МВ
Согласно международным согласительным документам [9] при организации специализированных центров муковисицидоза и протоколам ведения больных, в каждом центре штат сотрудников, должен включать кинезитерапевта, диетолога, психолога. Кинезитерапия является основополагающим методом лечения заболевания, не возможно обеспечить пациенту эффективную эрадикацию патогенов и сохранить легочную функцию без адекватного выведения мокроты. На практике, во многих центрах муковисцидоза специалисты, обладающие достаточными знаниями по данной патологии отсутствуют. Такая же проблема возникает и с врачами диетологами. Энергетическая потребность у пациентов с муковисцидозом составляет 150% от возрастной нормы, имеются особенности по введению прикорма, количеству потребляемой белковой нагрузки, пищевому поведению при развитии сахарного диабета (в отличие от остальных пациентов, у пациентов с МВ-зависимым сахарным диабетом углеводы не ограничиваются). Психологическая поддержка крайне важна для семей при постановке и принятии диагноза, в подростковом возрасте, в сложные для пациентов и их родителей периоды.
Пути решения организации помощи больным муковисицидозом в специализированных центрах
1. Организовывая специализированные центры муковисцидоза как самостоятельную единицу или на базе отделения стационара или КДЦ, необходимо исходить из рекомендаций Европейского общества [9]. Для 50 больных и более необходимо создание специализированного центра. Акцент делать на организации амбулаторной помощи. Подразделения стационара выполняют консультативно – диагностическую и лечебную помощь.
2. Штат сотрудников центра включает: пульмонолога, врача функциональной диагностики, кинезитерапевта, диетолога, психолога, должны быть организованы консультации гастроэнтеролога, лор – врача, эндокринолога, вакцинолога, аллерголога, трансплантолога и др.
3. Должна быть организована школа для пациентов и их семей.
Пути решения проблем, связанных с отсутствием центра МВ
1.При отсутствии центра МВ должны быть назначены пульмонолог, гастроэнтеролог и диетолог, осуществляющие помощь больным МВ, которые направляются на специализированное обучение.
2. Каждый пациент должен быть информирован о необходимости ежедневного дренажа мокроты после ингаляции муколитиков методами кинезитерапии (включая применение дыхательных тренажеров и аппаратных методик), особенностях диеты и ферментной терапии, питьевого режима, приема соли для профилактики синдрома Псевдо – Баттера.
3. Семьи больных должны быть проинформированы о сайтах с достоверной информацией, должны быть организованы горячие линии для получения ответа на многочисленные вопросы.
Остановимся на двух общих актуальных проблемах оказания помощи больным МВ. Общей проблемой для пациентов РФ является обеспечение жизненно важными медицинскими изделиями (ингалятор, дыхательные тренажеры, аппараты для бронхолегочного дренажа) и лечебным питанием, не входящих в стандарт медицинской помощи. Решение данной проблемы возможно при наличии медицинских показаний по решению врачебной комиссии (статья 37 п.5 ФЗ от 21.11.2011 N 323-ФЗ (ред. от 29.12.2015) «Об основах охраны здоровья граждан в Российской Федерации»).
Не организована психологическая поддержка пациентов с МВ, их семей. Решить эту проблему частично может создание возможности контакта между пациентами и их семьями за счет создания и развития интернет-площадки для общения с привлечением профильных специалистов медиков и психологов, а так же посредством «горячей линии».
Спектр существующих в настоящее время проблем у пациентов с муковисицидозом многогранен, но при своевременном их решении, возможно значительно увеличить продолжительность и качество жизни пациентов, осуществлять методы профилактики заболеваемости в стране и значительно сократить затраты на лечение и реабилитацию данной категории больных.
Список литературы:
1. Cystic Fibrosis Foundation Patient Registry 2014 Annual Data Report
2. Dodge, J.A., Lewis, P.A., Stanton, M., and Wilsher, J. «Cystic fibrosis mortality and survival in the UK: 1947–2003». Eur Respir. 2007; 29: 522–526
3. Капранов Н.И., Каширская Н.Ю. Монография «Муковисцидоз» Медпрактика 2014г
4. Infection Control Guidance for Patients with Cystic Fibrosis Ratified by Infection Control & Decontamination Assurance Group: 8th April 2014
5. Блистинова З.А. «Клиническое значение стационарозамещающих технологий при лечении реабилитации и медико-социальной адаптации больных муковисцидозом» диссертация кандидата медицинских наук,Москва, 2002г
6. «Регистр больных муковисицидозом в Российской Федерации.2013г» ИД «Медпрактика-М»,2015,64с
7.Данные о результатах исследования http://mukoviscidoz.org/docs.html
8. Chuchalin A, Kapranov N et al, Pediatr Drugs 2007; 9 Suppl. 1: 21
9. Smyth AR1, Bell SC2, Bojcin S3, Bryon M4, Duff A5, Flume P6, Kashirskaya N7, Munck A8, Ratjen F9, Schwarzenberg SJ10, Sermet-Gaudelus I11, Southern KW12, Taccetti G13, Ullrich G14, Wolfe S15; «European Cystic Fibrosis Society. European Cystic Fibrosis Society Standards of Care: Best Practice guidelines», J Cyst Fibros. 2014 May;13 Suppl 1:S23-42. doi: 10.1016/j.jcf.2014.03.010.